
Abstract
Background
Neurofibromatosis type 1 (NF1) is a multisystem genetic disorder with autosomal dominant inheritance which predisposes the affected individuals to increased risk of develo** certain benign and malignant central nervous system (CNS) tumors. NF1 patients are most notably prone to develop low-grade optic pathway, brainstem, and cerebellar astrocytoma. Current literature suggests that brain tumors in patients with NF1 tend to be less aggressive compared to sporadic ones. Glioblastoma multiforme (GBM) is a high-grade glioma which is relatively rare in patients with NF1 and is most commonly seen in supratentorial regions of the brain.
Case presentation
A 33-year-old patient was admitted in neurosurgery ward with acute hydrocephalus caused by a cerebellar mass lesion. On primary assessment, the patient was diagnosed with NF1. He was followed for 2 months and underwent surgical resection of the mass due to worsening symptoms. The pathology report revealed the malignant nature of the lesion. Patient received adjuvant chemo-radiotherapy with diagnosis of cerebellar GBM. Up to 19 months following surgery, he had gained a relatively well ability to walk and talk again.
Similar content being viewed by others

Background
Neurofibromatosis type 1 (NF1) is a common neuro-cutaneous condition with an approximate incidence of 1 in 3500 individuals in the general population [1]. The disease has a variable clinical course and is characterized by café-au-lait spots, freckling of skin folds, skeletal abnormalities, Lisch nodules and the tendency to develop certain benign and malignant neoplasms, most notably of the central nervous system (CNS) [2]. The majority of brain tumors in NF1 patients are low-grade gliomas and tend to have a more indolent course compared to the general population. Therefore, physicians follow brain tumors in NF1 patients conservatively [3].
Recent reports have warned neurosurgeons that gliomas in NF1 patients do not always have a benign histologic grade. With the help of immunohistochemical (IHC) staining further instances of higher grade lesions are being reported, changing the previous concept that “most brain lesions in NF1 have a low-grade pathology, a benign behavior and rarely do they transform to malignant tumors”. Comparing brain tumors in NF1 patients to their same site counterparts in the general population has shown a higher frequency of malignancy in NF1 patients [4]. Also, the risk of develo** malignant gliomas in NF1 patients is estimated to be fivefold compared to non-NF1 individuals [5].
Regarding anatomical location of high-grade gliomas in the general population, infratentorial location is quite rare compared to supratentorial. Accordingly, brain stem and cerebellar gliomas merely encompass 4.4% of adult gliomas [6].
Herein, a 33-year-old NF1 patient diagnosed with glioblastoma multiforme (GBM) tumor occurring in the posterior cranial fossa is reported, with some points regarding management strategies, clinical outcome, and a review of literature provided.
Case presentation
Presentation
A 33-year-old male patient presented to the emergency department, complaining of progressive headache, dysarthria, tremor, and ataxia. In the primary assessment, numerous café-au-lait spots and multiple neurofibromas were noticed in the patient (Fig. 1), raising clinical suspicion of an underlying neuro-cutaneous disorder. Neither he nor any of his family members were previously diagnosed with neurofibromatosis. The patient had no history of any significant neoplasm in his first-degree family members. Later by history taking, he stated that his father and two of his brothers had similar skin lesions for which they had not sought medical consultation.

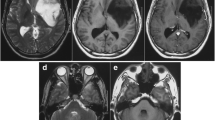
Patient’s physical exam revealed a multiple neurofibromas, b café-au-lait spots, and c axillary freckling
At the time of presentation, the patient had a decreased level of consciousness with a Glasgow Coma Scale of 13. Neuroimaging with CT scan (General Electric Healthcare, BrightSpeed, 16 slice CT scanner, USA, Illinois) was indicative of acute hydrocephalus caused by a posterior cranial fossa mass lesion, distorting and obstructing the 4th ventricle (Fig. 2a, b). Due to an acute decrease in the level of consciousness and the emergent need of surgical intervention and unavailability of endoscopic third ventriculostomy (ETV) setup, the patient underwent emergent surgical right anterior ventriculo-peritoneal (VP) shunting procedure (Fig. 2c, d). During the first post-operative day, he regained normal state of consciousness. His neurologic evaluation showed positive Romberg test and impaired finger to nose on the left side. A brain MRI (General Electric Healthcare, SIGNA HDx 1.5 Tesla, USA, Illinois) was demanded which revealed two gadolinium-enhanced lesions in the left cerebellar hemisphere, the smaller one in a deep location behind the left tectal plate and the larger one in a more cortical location below the tentorium (Fig. 3).

a, b Initial CT scans showed a posterior cranial fossa mass obstructing the 4th ventricle. c, d The hydrocephalus was resolved with a ventriculo-peritoneal shunting procedure

a–c T1-weighted axial and sagittal MRI images showing two mass lesions in the left cerebellar hemisphere which show enhancement after gadolinium injection and d T2-weighted axial MR image showing severe peri-tumoral edema, all in favor of GBM
Decision for surgical intervention
Assuming that the mass was probably of benign nature, patient’s management plan was close follow-up with neurologic exam and neuroimaging. Six weeks later, he developed aspiration pneumonia due to a weakened gag reflex. He was admitted in the Internal Medicine Ward and received a 7-day course of intravenous 600 mg Clindamycin every 8 h plus 1 gr intravenous ceftriaxone every 12 h combined with supportive respiratory care. After complete recovery from pneumonia, patient’s management plan was set to undergo elective surgical resection 2 months following the initial diagnosis. Microscopic total resection of the larger lesion was performed through a midline sub-occipital craniotomy and the tissue was sent for pathologic examination. The smaller lesion was not approached due to proximity to brain stem. In post-operation neurologic assessment, the patient had absence of gag reflex on the left side. Consequently, he underwent tracheostomy tube insertion and was discharged. Three weeks later, the tracheostomy tube was removed.
Histological evaluation
Pathology report of the tumoral tissue showed hypercellularity with neurofibrillary background and areas of microcytic pattern with vascular proliferation and foci of necrosis in favor of GBM. Tissue cells showed pleomorphism, atypia, and hyperchromasia. Mitosis was 1-2/Hpf. The lesion was positive for glial fibrillary acidic protein (GFAP), Oligo-2, and S100 on IHC analysis. Proliferation marker Ki67 was roughly positive in 5% of cells and no P53 positive cells were found in the tumoral tissue. The cells were also negative for CD-34 and IDH-1 (Fig. 4).

Sections show an ill-defined relatively hyper cellular mass with neurofibrillary background (a, b × 40, hematoxillin and eosin) with areas of necrosis (c, arrow, × 40, hematoxillin and eosin) and microvascular proliferation (d, arrow, × 40, hematoxillin and eosin). Immunohistochemical evaluation shows diffuse positivity for GFAP (e, × 400) with low (about 5%) Ki-67 positivity (f, × 400) and high expression of Olig-2 (g, × 400)
Follow-up
Upon recovery from surgery, the chemotherapeutic agent temozolamide was concomitantly administered with radiotherapy. A total dose of 60 Gy in 30 fractions was delivered as adjuvant radiation therapy. In the most recent follow-up which was 19 months after surgery, he had a modified Rankin Scale (mRS) of 1 and a Glasgow Outcome Score (GOS) of 4 with Karnofsky Performance Score (KPS) of 90/100. The most recent MRI (19 months after the initial diagnosis) showed a slight decrease in size of the smaller lesion with complete resolution of the larger lesion (Fig. 5).

The most recent T1-weighted MRI showing a gadolinium-enhanced mass within the left cerebellar hemisphere in axial, sagittal, and coronal views (a–c, respectively). The mass was enhanced after gadolinium. d Axial T2-weighted most recent MR images with significant peripheral edema and compression on 4th ventricle
Discussion and evaluations
Neurofibromatosis type 1, also known as von Recklinghausen disease, is the most common type of neuro-cutaneous disorders. Half the patients inherit the disease in an autosomal dominant (AD) manner and the other half are the result of sporadic mutations in the NF1 gene, a tumor-suppressor gene located on chromosome 17 [7]. Neurofibromin, the product of NF1 gene, is expressed in all cell types but has highest concentrations in glial cells, neurons, leukocytes, and Schwann cells [8]. This protein belongs to the GTPase activating protein family which inactivates the RAS signaling pathway by hydrolyzing the attached GTP to RAS protein. Any production of malfunctioned neurofibromin or its decreased production results in impaired RAS oncogene inhibition. Increased RAS activity leads to uncontrolled cell growth [9]. The first dysfunctional NF1 allele is inherited as a germline mutation and an acquired mutation in the second allele during a somatic event leads to development of neoplasms. This loss of heterozygosity occurs during somatic rearrangement, recombination and deletion which may affect other genes located on chromosome 17 including P53, epidermal growth factor 2 (HER2), topoisomerase II alpha (TOP2A) and breast cancer gene 1 (BRCA1). Alongside NF1, other mutated genes collaborate in pathogenesis of neoplasms, most notably of the nervous system [10]. Among the nervous system neoplasms, NF1 patients are particularly prone to malignant peripheral nerve sheath tumors (MPNST), optic pathway gliomas and pilocytic astrocytomas [11]. Comparing to the general population, NF1 patients are at an increased risk of malignant gliomas as well benign ones [5].The risk of malignancy in NF1 patients has been reported to be 5 to 29% in several studies [12,13,14].
Herein, we present a case of NF1 diagnosed with multifocal left cerebellar GBM. GBM is the most common primary malignant brain tumor and the most aggressive type among gliomas with less than 5% survival rate beyond 36 months [15]. The majority of GBM patients develop the neoplasm sporadically; however, patients with disorders such as neurofibromatosis, Turcot syndrome, and Li–Fraumeni syndrome are genetically predisposed to sustain GBM [16]. Common molecular alterations in GBM include increased copies of EGFR gene, mutated P53 gene, inactivation of RB gene, and mutations in IDH-1 gene alongside other mutations [17, 18]. Abnormal NF1 gene, either mutated or deleted, is frequent in human GBM, as 23% of GBMs were reported to harbor aberrations in the NF1 gene [19]. Although NF1 patients usually present with low-grade gliomas, a handful of GBM tumors associated with NF1 have been reported.
Pál and colleagues, in 2001, reported a 37-year-old NF1 patient who died of progressive multiple sclerosis whose autopsy revealed a right hemisphere glioblastoma tumor which was not symptomatic during her life [20]. In another article in 2008, a 28-year-old NF1 patient diagnosed with lobar cystic GBM was presented. The patient was managed with surgical resection and adjuvant chemo-radiation therapy with 41 months survival [21]. Huttner and colleagues analyzed the molecular biology beyond GBM tumors in five NF1 pediatric patients. All five tumors demonstrated P53 mutation and increased EGFR copy numbers. The study suggested a more favorable prognosis could be expected for GBM tumors in NF1 patients rather than the sporadic cases [22]. In another case report of a 32-year-old NF1 patient with GBM, the tumoral cells were strongly positive for GFAP and negative for EGFR. The patient was treated with surgical removal of the tumor and adjuvant chemo-radiation. He had no remarkable symptoms and tumor recurrence up to 9 months post-operation follow-up [23]. In another article, a 9-year-old NF1 patient was reported who died of GBM 3 days following initial diagnosis, warning physicians to follow tumors in NF1 patients closely [24].
Shibahara and colleagues reported a unique subset of GBM in four NF1 patients. None of the patients had mutations in isocitrate dehydrogenase 1 (IDH-1) gene, v-RAF murine sarcoma viral oncogene homolog B1 (BRAF) gene, and telomerase reverse transcriptase (TERT) gene promoter [25].
Cerebral hemispheres are the most frequent sites of GBM, while less than 5% of cases are located in infratentorial regions of the brain [6]. We reviewed the current literature regarding infratentorial GBM tumors in NF1 patients. Few instances of posterior fossa GBM in NF1 patients has been reported up to the year 2019. A 28-year-old NF1 patient was followed for a cerebellar mass believed to be a hamartoma. Later, due to worsened symptoms, the mass was resected and pathology reported two distinct tumors attached together, a neurofibromas and a GBM. On IHC staining, the first lesion was positive for S-100 mutation and the latter was positive for GFAP and 30% of cells harbored P53 mutation. The study suggested close follow-up of brain lesions in NF1 patients for potential development of high-grade gliomas. The Patient received standard surgical and chemo-radiational therapy but showed signs of tumor metastasis in right frontal lobe and died 6 months after the treatment [26]. In another report from India, a 6-year-old NF1 patient was diagnosed with cerebellar GBM and despite surgical intervention and chemo-radiotherapy died after 4 months [27].
The patient we presented in the current study was diagnosed with infratentorial GBM which is a relatively rare site to expect GBM. In the general population, supratentorial GBM is more frequent than the infratentorial ones, so we hypothesize the rarity of infratentorial GBM in NF1 patients might be secondary to this fact. Interestingly, the presented tumor had unique molecular findings including the lack of IDH-1 and P53 mutation with positive Ki67 in 5% of cells and low mitosis activity (1-2/Hpf). Due to the tumor’s low mitosis activity, we expected a relatively favorable outcome, as in a previous meta-analysis, low Ki67 positivity was the strongest factor associated with a better prognosis [28]. Accordingly, in the most recent evaluation which was 19 months after initial diagnosis, the patient had an acceptable performance score and the most recent brain MRI showed a slight decrease in size of the residual tumor and no recurrence of the resected one.
Despite the common concept of high probability of benign nature of brain tumors in NF1 patients, though rare, possibility of malignant gliomas in NF1 patients should be kept in mind. Most gliomas in NF1 patients follow an indolent clinical course. On the other hand, they are at an increased risk of develo** malignancies; therefore we suggest close follow-up of brain mass lesions in NF1 patients and eminent surgical resection of lesions which grow quickly or induce neurological deterioration.
Availability of data and materials
Not applicable
Abbreviations
- CNS:
-
Central nervous system
- ETV:
-
Endoscopic third ventriculostomy
- GBM:
-
Glioblastoma multiforme
- NF1:
-
Neurofibromatosis type 1
References
Pong WW, Gutmann DH. The ecology of brain tumors: lessons learned from neurofibromatosis-1. Oncogene. 2011;30(10):1135–46.
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3):608–14.
Korf BR. Malignancy in neurofibromatosis type 1. Oncologist. 2000;5(6):477–85.
Ilgren EB. Gliomas in neurofibromatosis: a series of 89 cases with evidence for enhanced malignancy in associated cerebellar astrocytomas. Pathol Annu. 1985;20(1):331–58.
Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using US death certificates. Am J Hum Genet. 2001;68(5):1110–8.
Strauss I, Jonas-Kimchi T, Bokstein F, Blumenthal D, Roth J, Sitt R, et al. Gliomas of the posterior fossa in adults. J Neurooncol. 2013;115(3):401–9.
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152(2):327–32.
Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8(3):415–28.
Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12(1):1–11.
Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, et al. The NF1 gene revisited–from bench to bedside. Oncotarget. 2014;5(15):5873–92.
Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. 1986;314(16):1010–5.
Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis: a clinical and population study in south-east Wales. Brain. 1988;111(6):1355–81.
Hope DG. Malignancy in neurofibromatosis. Adv Neurol. 1981;29:33–56.
Brasfield RD, Gupta TD. Von Recklinghausen’s disease: a clinicopathological study. Ann Surg. 1972;175(1):86–104.
Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130(10):2596–606.
Alifieris C, Trafalis DT. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82.
Sasmita AO, Wong YP, Ling AP. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia Pac Clin Oncol. 2018;14(1):40–51.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Pal E, Gömöri É, Gáti I. Neurofibromatosis and glioblastoma in a case of multiple sclerosis. Eur J Neurol. 2001;8(6):717–8.
Hakan T, Aker FV. Case report on a patient with neurofibromatosis type 1 and a frontal cystic glioblastoma. Neurol Neurochir Pol. 2008;42(4):362–5.
Huttner AJ, Kieran MW, Yao X, Cruz L, Ladner J, Quayle K, et al. Clinicopathologic study of glioblastoma in children with neurofibromatosis type 1. Pediatr Blood Cancer. 2010;54(7):890–6.
Jeong TS, Yee GT. Glioblastoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Brain Tumor Res Treat. 2014;2(1):36–8.
Distelmaier F, Fahsold R, Reifenberger G, Messing-Juenger M, Schaper J, Schneider DT, et al. Fatal glioblastoma multiforme in a patient with neurofibromatosis type I: the dilemma of systematic medical follow-up. Childs Nerv Syst. 2007;23(3):343–7.
Shibahara I, Sonoda Y, Suzuki H, Mayama A, Kanamori M, Saito R, et al. Glioblastoma in neurofibromatosis 1 patients without IDH1, BRAF V600E, and TERT promoter mutations. Brain Tumor Pathol. 2018;35(1):10–8.
Broekman ML, Risselada R, Engelen-Lee J, Spliet WG, Verweij BH. Glioblastoma multiforme in the posterior cranial fossa in a patient with neurofibromatosis type I. Case Rep Med. 2009;2009:757898.
Incecik F, Hergüner MO, Bayram I, Zorludemir S, Altunbasak S. Fatal glioblastoma multiforme in a child with neurofibromatosis type 1. Indian J Cancer. 2015;52(3):298–9.
Reavey-Cantwell JF, Haroun RI, Zahurak M, Clatterbuck RE, Parker RJ, Mehta R, et al. The prognostic value of tumor markers in patients with glioblastoma multiforme: analysis of 32 patients and review of the literature. J Neurooncol. 2001;55(3):195–204.
Acknowledgements
Not applicable
Funding
None
Author information
Authors and Affiliations
Contributions
NA conceived the study and wrote the manuscript. ND assisted in the preparation of the manuscript. All authors critically reviewed the manuscript and approved the final version of the manuscript and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Derakhshan, N., Azadeh, N., Saffarian, A. et al. Cerebellar glioblastoma multiforme in an adult patient with neurofibromatosis type 1: an extremely rare report with review of literature. Egypt J Neurol Psychiatry Neurosurg 55, 85 (2019). https://doi.org/10.1186/s41983-019-0135-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-019-0135-2