
Abstract
Background
Exosomes are extracellular vesicles secreted by eukaryotic cells and have been extensively studied for their surface markers and internal cargo with unique functions. A deeper understanding of exosomes has allowed their application in various research areas, particularly in diagnostics and therapy.
Main body
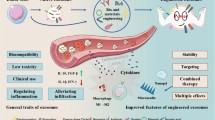
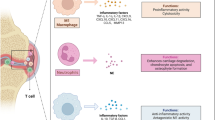
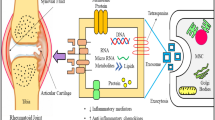
Exosomes have great potential as biomarkers and delivery vehicles for encapsulating therapeutic cargo. However, the limitations of bare exosomes, such as rapid phagocytic clearance and non-specific biodistribution after injection, pose significant challenges to their application as drug delivery systems. This review focuses on exosome-based drug delivery for treating rheumatoid arthritis, emphasizing pre/post-engineering approaches to overcome these challenges.
Conclusion
This review will serve as an essential resource for future studies to develop novel exosome-based therapeutic approaches for rheumatoid arthritis. Overall, the review highlights the potential of exosomes as a promising therapeutic approach for rheumatoid arthritis treatment.
Graphical Abstract

Similar content being viewed by others
Background
Exosomes are extracellular vesicles of 30–100 nm diameter and are secreted by most eukaryotic cells [1]. They are secreted from various types of cells and contribute to homeostasis by participating in intercellular interactions [2,3,4]. Conventionally, exosome research has focused on surface markers or their internally encapsulated cargo. CD9 and CD63 are verified exosome-specific markers [5]. In addition, the components of the internal cargo (lipids, nucleic acids, metabolites) have been characterized [5, RA is a chronic disease that causes severe pain, swelling, stiffness, and functional loss of joints [17,18,19]. Although the exact pathogenesis of this disease is unknown, RA onset is reportedly caused by various risk factors, including genetic, epigenetic, dust inhalation, microbiota, and lifestyle [20,21,22]. Immune regulatory factors related to nuclear factor kappa B (NF-κB) stimulation, activation, and functional differentiation of T cells are believed to be closely related to RA. Human leukocyte antigen (HLA)-DBR1 reportedly has the most robust relation with RA among genetic factors. The effects of environmental factors are apparent when investigated at the genetic level. For example, smoking and silica inhalation significantly increase the incidence rate of RA in individuals with susceptibility to HLA-DR4 alleles. Although RA is a systemic autoimmune disease, symptoms usually occur with severe joint inflammation [23]. Thus, targeting the inflammatory disease microenvironment and cell populations is crucial for treating RA [24]. In synovial tissue, highly proliferative immune cells, vascular endothelial cells, and fibroblast-like synoviocytes (FLSs) establish an inflammatory microenvironment and release various factors such as TNF-α, IL-1, IL-6, matrix metalloproteinases (MMPs), and autoantibodies [25,26,27,28,29]. These components facilitate the development of hyperinflammatory microenvironment. In addition, systemic impaired immune functions and an imbalanced proportion of immune cells, such as in the helper T cell and regulatory T cell ratio, further aggravates RA pathology. The cell types closely related to RA pathogenesis include FLSs, macrophages, B cells, T cells, and dendritic cells (DCs) (Fig. 1). A The modulation of various kinds of cells in rheumatoid arthritis-microenvironment by engineered exosomes. B Immunosuppression effects on macrophages, dendritic cells (DCs), and T cells. FLS, fibroblast-like synoviocytes; Th1, T-helper 1 cells; Th17, T-helper 17 cells; Treg, Regulatory T cells FLS, a significant producer of inflammatory cytokines at the synovial site, is primarily responsible for RA initiation [30, 31]. FLSs constitute a 2–3-layer network in normal synovium. However, in the RA pathological condition, an inflammatory response is induced by continuous FLS proliferation and immune cell accumulation, which causes synovial hyperplasia [32]. Activated FLS converts its network to a pannus-like multi-layer structure in RA lesions. This structure can be identified as a distinct synovium lining hypertrophic lesion [33]. During RA progression, MMP-13 overexpression mediates the destruction of bone and cartilage structures. Multiple studies have reported that MMP-13 production is increased in cytokine-stimulated FLSs [34, 35]. Activated FLSs act as effector cells that induce the activation of inflammatory T cells by expressing IL-7 and IL-15, which possess functional similarities with IL-2 [36, 37]. However, synovial macrophages secrete only IL-15, whereas synovial T cells do not secrete either IL-7 or IL-15. In addition, FLS secrete TNF-α similar to other synovial cells such as lymphocytes, macrophages, and endothelial cells [38]. Therefore, cytokines secreted by synovial immune cells and the distinct cytokine profile of FLS contribute to the RA microenvironment. Activated macrophages secrete IL-1, IL-6, and TNF-α, and they can be induced by various cell subsets, such as FLSs, monocytes, and helper T cells [39]. The cytokines present in the synovial fluid induce severe inflammation in RA [40, 41]. Initial erosion of RA tissue gives way to synovial hyperplasia [42, 43]. Activated macrophages predominantly accumulate at the borderline of pannus-like multi-layer structure and cartilage along with FLSs [33]. In particular, TNF-α and IL-1 induce FLS activation, producing tissue-destructive MMP [44]. TNF-α inhibition by antibodies leads to a considerable reduction in IL-1, IL-6, IL-8, and GM-CSF. Therefore, TNF-α blockade is an excellent therapeutic strategy to suppress inflammation in RA patients. CD4+ T cells are considered central cellular mediators involved in RA pathogenesis [45]. Among CD4+ T cells, helper T cells and regulatory T cells are significantly related to RA. Helper T cells, characterized by CD4 expression on the cell surface, promote inflammation by activating osteoclasts, FLSs, chondrocytes, and immune cells via various inflammatory cytokines. For example, helper T cells secrete IL-17 to activate osteoclast, which causes bone destruction [46]. Similarly, IFN-γ contributes to macrophage polarization to inflammatory M1 phenotype [47]. In addition, cell surface signaling by membrane proteins such as CD69 and CD11 induces the secretion of MMP and other effector molecules by FLS and chondrocytes, which in turn promotes inflammatory responses and causes joint erosion [23]. Another subset of CD4+ T cells is regulatory T cells (Tregs), CD4+CD25+FOXP3+ cells [48]. Unlike helper T cells, Tregs are considered effective suppressors of RA [49]. Although many Treg cells are present in the synovial fluid of RA patients, they are functionally deficient. This suggests an immune suppressive role of ‘normal’ Tregs in RA [50]. Thus, synovial Tregs in RA patients often exhibit reduced regulatory capacity, which might be attributed to the severe inflammatory condition of the RA microenvironment [51]. DCs are one of the most professional antigen-presenting cells (APCs) [52]. DCs prime T cells by presenting antigenic peptides on their surface complexed with MHC [53]. Along with MHC class II, co-stimulatory molecules such as CD80 and CD86 facilitate interaction and priming of T cells [54]. Auto-reactive helper T cells primed by DCs contribute to the inflammatory microenvironment by secreting pro-inflammatory cytokines, such as IL-12 and IL-23 [55]. Like other immune cells, B cells are also abnormally increased in RA [56]. During RA initiation, B cells produce autoantigen, TNF-α, and IL-6 [57]. Among them, IL-6 contributes to forming an inflammatory microenvironment in the synovial membrane by promoting monocyte differentiation. In addition, B cells act as APCs in RA to induce helper T cell activation by presenting autoantigens to CD4+ T cells [58]. As the disease progress, B cells infiltrate synovial tissue and induce a sustained and destructive autoimmune response. The synovial microenvironment facilitates optimal antigen processing and presentation by antigen-presenting cells for T cell activation [59]. Studies have revealed that the interaction of activated T cells with monocytes induces the secretion of various inflammatory cytokines and chemokines [60,61,62]. Hence, the interaction of immune cells and inflammatory factors in the inflamed synovial microenvironment plays a pivotal role in RA pathogenesis and progression. Exosomes are nano-sized extracellular vesicles secreted by most eukaryotic cells. Unlike other small extracellular vesicles such as apoptotic bodies and microvesicles, exosomes are primarily generated via cell membrane fusion of exosome-containing endosomes. Exosomes typically have a hydrodynamic size of 30–150 nm in diameter, which can be characterized via nanoparticle trafficking analysis method. The physicochemical properties of exosomes can be characterized through isolation from conditioned cell culture media using ultracentrifugation, size exclusion chromatography, or immunoaffinity-based isolation kits [63]. Exosomes express CD9 and CD63 as surface markers and encapsulate exosome biogenesis-related factors, such as TSG101, ALIX, and Rab27a. Various studies have recently studied exosome biogenesis, and the accumulated research results provide evidence about exosome biogenesis [1, 64, 65]. Exosomes are generated via direct inward budding of the phospholipid bilayer within the MVB derived from the endosomes and then secreted out of the cell [64]. Exosome secretion occurs through an endosomal sorting complex responsible for transport (ESCRT) protein-dependent or ESCRT-independent mechanism. ESCRT-independent exosome biogenesis showed that sphingolipid ceramide is crucial [66]. This study has revealed that the inhibition of neutral sphingomyelinases reduces the number of exosomes released. In addition, cells can produce CD63-positive MVEs regardless of the depletion of the four subunits of ESCRT [67]. However, several ESCRT proteins are involved in exosome formation and induce ESCRT-dependent exosome biogenesis. For example, the reticulocyte transferrin receptor reportedly interacts with ESCRT accessory protein Alix during exosome formation [68]. Subsequently, Alix contributes to exosome biogenesis and intraluminal vesicle formation by recruiting ESCRT-III complex protein [69]. Various mechanisms determine the contents of the cargo inside the exosome, and as a representative example, proteins such as tetraspanin participate in protein loading in the exosome. Tetraspanin-rich microdomains contribute to the differentiation of receptors and signaling molecules within the exosome plasma membrane [70]. In addition, tetraspanins such as CD9 and CD81 are known to form microdomains and bind to specific proteins; this function serves as a sorting mechanism during the generation of exosomes [70, 71]. Rab27 is an essential protein involved in exosome secretion after biogenesis. Rab27 silencing reduces exosome secretion without changing exosome cargo protein composition [72]. Conversely, inhibiting the proteasomal degradation of Rab27 tends to increase exosome secretion [73]. Various cargo molecules, including DNAs, RNAs, and lipids, are present inside the exosome [74]. Cargos are considered to play an essential role in intercellular communication. In contrast to the previous assumption that exosomes are just cell debris, recent studies have revealed that they are primarily involved in normal biological processes [75]. The exosomes mediate local communication around the recipient cell, and they circulate along the blood vessel after being secreted and accumulate at the target site, acting as mediators in cell-to-cell interactions [76, 77]. Exosomes delivered to the recipient cell enter the recipient cell through fusion, receptor-mediated uptake, and internalization for cell-to-cell communication. A previous study reported that mRNA could be encapsulated in the exosome of the parent cell and translated after being taken up by the recipient cell [7]. Studies on the development of delivery systems have explored the applicability of exosomes to treat diseases [78]. Previous exosome studies reveal that inhibiting exosome biogenesis induces cell apoptosis [64]. Many studies have revealed that exosomes play a crucial role in the progression of intractable diseases, and efforts to understand the exosomes related to the diseases are continuing. In RA, exosomes significantly contribute to the inflammatory environment of the joints. Numerous exosomes exist in various biological fluids, including synovial fluid [79]. These exosomes are secreted from multiple cell populations such as FLSs, T cells, and B cells [80,81,82,83]. They trigger inflammation by delivering damage-associated molecular patterns [84]. In addition, the exosome’s cytokines, lipids, and microRNAs aggravate RA’s inflammation [85]. Exosomes also play critical roles in disease progression in various chronic diseases. For example, cancer exosomes deliver oncogenic cargos, which makes tumors a favorable microenvironment. They also overexpress PD-L1 on their surface, thereby resisting immune checkpoint inhibitors. Disease progression can be delayed, and the therapeutic effect of conventional therapies can be improved by targeting disease-favorable exosomes. Therefore, exosomes are potential biomarkers in chronic diseases and have the potential to develop therapeutic approaches [86]. Growth in the knowledge of the intrinsic biological functions of exosomes has led to the development of various exosome engineering strategies with biomedical applications. Considering these inherent biological functions of exosomes, bare-state exosomes from various types of cells were utilized to treat intractable diseases, including cancer, RA, multiple sclerosis, and diabetes [87,88,89]. Although exosomes have emerged as a replacement for cell-based therapy owing to their remarkable therapeutic efficacy, the regulation of exosomal compartments has been attempted to maximize their effects. Although exosomes possess homing effect, which can be recruited to parent cells, several engineering strategies have been proposed that disadvantages such as low targetability and poor distribution have still been issued. The engineering strategies can be classified into two categories: modification before and after the isolation of exosomes (Fig. 2). Exosome engineering via pre-isolation modification and post-isolation modification Exosomes can achieve the distinct properties of parent cells by loading the same protein, RNA, and DNA with origin cells as well as by expressing the membrane protein of secreted cells in the exosomal surface [1]. Accordingly, based on exosome biogenesis, the engineering methods for modulating parent cells are developed. The focus of the research includes pre-conditioning methods for altering cell characteristics using various cytokines or stimuli, endogenous modulation for regulating genetic parts, cell membrane modification for exosomal membrane surface engineering, and pre-treatment for cargo modification via the utilization of exosome biogenesis pathway (Fig. 3). Schematic illustration of pre-isolation modification methods. A Exosomes from target ligand (avidin) labeled and anti-tumor drugs encapsulated donor cells for further labeling with avidin complex [94], as therapeutics for remyelination [95], and migraine [96]; however, tolerogenic Dex are used in the treatment of several autoimmune diseases, including multiple sclerosis [106]. Unlike M1-derived exosomes, M2-derived exosomes facilitate the reprogramming of M1 to M2, reflecting anti-inflammatory properties. Owing to intrinsic properties similar to that of M2, M2-derived exosomes are used as therapeutics for various diseases, such as RA, wound healing [116]. Theranostic nanoparticles and imaging probes such as fluorescent dyes were labeled onto the azide (N3) functional group via copper-free click chemistry utilizing metabolic glycoengineering using mannosamine. Although this strategy was mainly applied for in vivo tracking of several types of cells [117,118,119] and active-targeting of tumors [120], metabolic glycoengineering or bioorthogonal click chemistry was recently used for in situ one-step strategy to label exosomes, considering the exosome biogenesis pathway [93]. The authors noted that the yield of acquisition of dye-labeled exosomes from synthetic metabolite-treated donor cells was higher than that by applying bioorthogonal click chemistry directly to isolated exosomes, and also it preserved the intrinsic properties of exosomes. In addition to the fluorescent dye conjugation, a study of exosomal surface-modified exosomes with biocompatible polymer via bioorthogonal chemistry based on metabolic glycoengineering was conducted. Park and group reported PEGylated hyaluronic acid (HA)-coated exosomes to target the CD44-overexpressing cells actively and verified the targetability using the RA and PC3 tumor-bearing mouse models, representative models of CD44-overexpression [121]. Briefly, the authors induced the expression of the N3 group in the cellular membrane of donor cells by pre-treatment with N-azidoacetyl-D-mannosamine (Ac4ManNAz), and the parent cells utilized in this research were cancer cells regarding the proof of concept. Subsequently, dibenzocyclooctyne-terminated PEGylated-HA was conjugated with the N3 group using bioorthogonal copper-free click chemistry. Therefore, the PEGylated-HA functionalized exosomes were isolated using ultracentrifugation, and surface editing was verified using biolayer interferometry (BLI) and confocal imaging. Thereafter, macrophage-targeing dextran sulfate (DS)-coated adipose-derived stem cells-derived exosomes for immunomodulation in RA were suggested by the same group [122]. The strategy for exosomal surface editing has been described previously; however, DS was introduced to target the macrophage scavenger receptor class A (SR-A) of activated macrophages. Owing to the sophisticated physicochemical properties of phospholipid bilayer structures of cellular membranes, various functional moieties linked with lipid molecules can be incorporated into the cell membrane surfaces by hydrophobic insertion via physical interaction. Furthermore, this modification strategy can endow the exosomal membrane with functional moieties introduced into cells. Wang et al. amended the donor cells to obtain the dual ligand-modified and drug-encapsulated exosomes [123]. The authors pre-treated 1,2-distearoyl-sn-glycero-phosphoethanolamine (DSPE)-PEG-Biotin to introduce the biotin on cellular membrane surfaces, utilizing the physical interaction between DSPE and lipid bilayer of the cellular membrane. Subsequently, the cells were pre-treated with anti-tumor drugs, assuming that the drug will be packaged in exosomes following uptake into the cytosol. Although the anti-cancer drug paclitaxel (PTX) induced partial apoptosis, the authors noted that PTX was also well-encapsulated in exosomes. To edit the exosomes with dual ligands and enhance the targetability, Wang et al. subsequently treated donor cells with avidin, resulting in a biotin-avidin reaction, and successfully isolated these exosomes utilizing the microfluidic chips created by them. Wang et al. also applied the similar strategy to obtain biotinylated exosomes for further engineering with avidin modified with molecular beacon and poly dopamine (PDA) covered Fe3O4 nanoparticles [65], exosomes from various cells have been widely used to modify the surface of theranostic nanoparticles via diverse strategies such as sonication, extrusion, and incubation. The hydroxychloroquine (HCQ)-loaded zinc sulfide (ZnS) nanoparticles, which act as a photosensitizer, were coated with glioblastoma cells-derived exosomes through thin film hydration followed by extrusion and subsequently modified with iRGD peptide which provided the pH- and redox- responsiveness [142]. This study noted that, owing to the homing ability of exosomes, HCQ@ZnS@eRGD was permeable across the blood–brain barrier and showed enhanced targetability to glioblastoma cells in vivo. In addition to the inorganic nanoparticles, exosome-coated poly(lactic-co-glycolic acid) (PLGA) nanoparticles loaded with 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaine iodide (DiR) were obtained using microfluidic sonication and exhibited a prolonged blood circulation, suggesting the effects of exosomal surfaces [130]. Even though the endogenous modulation of origin cells facilitated the engineering of exosomal proteins and genes, they can be loaded into isolated exosomes by controlling the loading efficiency from the protein or gene delivery view. Wan and Zhong et al. reported a study related to tissue-specific gene therapy of liver fibrosis using Cas9 ribonucleoprotein (RNP) delivery exosomes [133]. The exosomes were isolated from LX-2 cells through ultracentrifugation, and Cas9 RNP complexes were loaded into exosomes via electroporation with 20% entrapment efficiency. Meanwhile, the human umbilical cord MSC-derived exosomes modified with polypeptide (CAQK peptide) and loaded with the CRISPR/Cas9 components were applied for the immunotherapy of spinal cord injury [143]. The CAQK peptide was chemically conjugated on exosomal surfaces via EDC/NHS chemistry, and CRISPR/Cas9 plasmid was loaded using electroporation methods. In addition to the electroporation strategy, McAndrews et al. used the commercially available transfection reagent, Exo-Fect Exosome Transfection Kit, to encapsulate the CRISPR/Cas9 plasmid DNA into MSC-derived exosomes for oncogenic Kras targeted therapy [152]. Even though several stimuli-sensitive drug delivery systems, which facilitate active and passive targeting, have been evaluated, their clinical application remains to be validated in various experimental and preclinical systems. Lee and Sul evaluated reactive oxygen species-responsive tolerogenic exosomes for RA treatment since ROS is overproduced in the inflamed joints [153]. Ma et al. explored chemical reaction-based novel surface-modification strategy, which can be used in situ to modify the ascorbic acid (AA), an antioxidant, on the exosomal surface to overcome the low loading efficacy of common loading methods [132]. In this study, mesenchymal stem cell-derived exosomes were isolated using ultracentrifugation and incubated with DSPE-PEG-SH followed by incubation with HAuCl4 solution to introduce Au3+ on exosomal surfaces. Because AA can act as a reducing and protective agent for AuNPs growth, AA solutions were incubated with Au3+-introduced exosomes. Subsequently, exosomes with their surface-modified with AA-protected AuNPs (mExo@AA) were obtained and used as eye drop formulation to treat dry eye disease. Its therapeutic effects were achieved by eliciting damage repair, ROS scavenging, and macrophage polarization into the M2 type. For gene/chemo/photothermal therapy and molecular imaging, a combination system composed of polydopamine (PDA) coated magnetic Fe3O4 nanoparticles and DOX-loaded exosomes was prepared using various exosome engineering strategies, including pre-isolation modification and affinity-based reaction [7) [170]. A Schematic illustration of post-isolation modification method for treatment of RA. A Preparation strategy of anti-ROS-CII antibodies loaded polymorphonuclear leukocytes (PMN)-derived extracellular vesicles (EVs). [170]. Reproduced with permission, Copyright 2020, Frontiers. B Schematic illustration of Evs-EGCG and cartilage regeneration. [171] Copyright 2021, Elsevier. C Schematic depicting of BBR-PEVs and RA microenvironment reprogramming [172]. Copyright 2021, Wiley The polymorphonuclear leukocyte (PMNs)-derived exosomes were isolated by differential centrifugation. Then the mixture of exosomes, dried phospholipids consisting of 1,2-dioleoyl-sn-glycero-3-phospho-L-serine, and antibodies was sonicated for 5 min with three amplitude settings. Several kinds of antibodies, including CII post-translationally modified by oxidants-specific antibody (anti-ROS-CII), viral IL-10 fused anti-ROS-CII (anti-ROS-CII-vIL-10), anti-mouse TNFα antibody (anti-mTNF), and both anti-ROS-CII-vIL-10/anti-mTNF (anti-ROS-CII&mTNF/vIL-10), were enriched in exosomes. The data showed that the anti-ROS-CII incorporated exosomes (EV/anti-ROS-CII) bound explicitly to articular cartilage and were highly retained in the arthritic joints without off-target effects compared to non-modified exosomes. Moreover, the binding ability to both ROS-CII and mTNF of anti-ROS-CII&mTNF/vIL-10 incorporated exosomes revealed that the sonication led to incorporation of more than one antibody. In AIA, the EV/anti-ROS-CII&mTNF/vIL-10, which accumulated in the inflamed knee joints after intravenous injection, attenuated the synovial inflammation by downregulating the genes involved in pro-inflammatory response and cartilage degradation, and upregulating the genes related to anti-inflammatory response and cartilage regeneration. Furthermore, histological analysis revealed that EV/anti-ROS-CII bound to the intermediated and deep cartilage showed chondroprotection effects correlating to the genomic response. The sonication method allows the small-sized therapeutic molecules to be loaded into exosomes. Song et al. encapsulated the epigallocatechin gallate (EGCG), a small antioxidant molecule, into macrophage-derived exosomes to improve its utility against chondrocytes in physiological conditions (Fig. 7B) [171]. The macrophage-derived exosomes were isolated from RAW cells and subsequently EGCG-loaded exosomes (EVs-EGCG) were obtained. Encapsulation and sustained-release behavior of EGCG were verified using UV–Vis absorbance. The results showed that EVs-EGCG internalized into chondrocytes, attenuated hypoxia-inducible factor 1-alpha (HIF-1α)-mediated inhibition of apoptosis, and augmented type II collagen expression, thereby alleviating RA symptoms in a rat model of CIA. The encapsulation of the small organic compounds can be made feasible using the incubation method [172, 173]. Ma et al. developed berberine, which is known to suppress the activated macrophages and DCs, -coordinated platelet-derived exosomes (BBR-PEVs) (Fig. 7C) [172]. The PEVs were isolated by centrifugation using ultrafiltration tubes (3 kD), and the incubation of DMSO-dissolved BBR was followed. The high-performance liquid chromatography (HPLC) data showed that the loading efficiency was 6.76% and that 91.9% of BBR was sustained-released within 48 h. The BBR-PEVs selectively accumulated into inflamed joints and reshaped RA microenvironments by reducing the proportion of T cells, macrophages, and DCs, downregulating the activated phenotypes of those cells, and regulating the cytokines to a normal state; these effects collectively resulted in relief of RA symptoms. He et al. loaded curcumin (Curc) into MSC-exosomes (Curc-Exos) using the incubation method [173]. The MSC-exosomes were isolated using Exocib kit, then and mixed with Curc solution. The incubated mixtures were centrifuged by sucrose gradient, and 45 and 60% layers were selected as Curc-Exos with 18 ± 5% loading capacity. The Curc-Exos exhibited acidic pH-triggered faster release, followed by apoptosis of HIG-82 cells, a synovial membrane cell line, and downregulated pro-inflammatory mRNAs, implying their anti-inflammatory effects on FLSs. Yan et al. utilized macrophage-derived exosomes as targeted drug delivery carriers [174]. They encapsulated dexamethasone sodium phosphate (Dex) into exosomes and modified the exosomal surface with folic acid-polyethylene glycol-cholesterol (FA-PEG-Chol) for active targeting of activated macrophages. The exosomes were isolated by differential centrifugation, and Dex was loaded (wt ratio of Exo: Dex = 1:3) using electroporation in the presence of 80 nM of trehalose to prevent aggregation. The electroporation was performed in a double poring pulse with 200 V for 5 ms and a transfer pulse of five pulses with 20 V for 50 ms at room temperature, followed by incubation in 37 ℃ PBS for 1 h to restore the exosomal membrane. Furthermore, the pre-synthesized FA-PEG-Chol was modified by post-insertion since because the cholesterol could be inserted into the lipid bilayer of the exosome. Exo/Dex and FA-PEG-Chol were mixed (wt ratio 1:5) and incubated at 37 ℃ for 2 h. The HPLC data showed the drug loading efficiency of Exo/Dex and FPC-Exo/Dex was 22.38% and 18.81%, respectively. The release of Dex was triggered by acidic pH, and the cellular uptake into activated macrophages was enhanced after surface modification, suggesting FA-mediated active targeting. In the CIA model, augmented accumulation of the FPC-Exo/Dex in inflamed joints suppressed inflammatory cell infiltration, pro-inflammatory cytokine release, as well as increase in anti-inflammatory cytokine level, which resulted in the alleviation of RA symptoms and mitigated bone erosion. The authors noted that FPC-Exo/Dex hardly manifested hepatotoxicity, suggesting their biocompatibility. Li et al. decorated the surface of M2 macrophage-derived exosomes (EXs) with a polyarginine peptide (R9) [178]. The developed exosome was considered for potential treatment of RA. The engineered exosome exhibited higher cellular uptake and showed an excellent targeting effect. For example, the engineered exosomes showed 176% accumulation at the site of inflammation. This was due to higher Cur@EXs-R9 capture and uptake by inflammatory cells. In addition, the M2 macrophage-derived exosome effectively repolarized the macrophage at the inflamed site, and the authors proposed that Cur@EXs-R9 is a potential treatment option for RA. Engineering strategies utilizing both pre-isolation and post-isolation modification have been introduced to confer multifunctionality on exosomes and maximize the therapeutic effects in RA. The parent cells are first engineered to exert immunosuppressive effects, and exosomes are isolated from these engineered cells and subjected to either exosomal surface modification or cargo loading. Lee et al. developed a reactive oxygen species (ROS), one of the RAM stimuli,-responsive tolerogenic dendritic cell-derived exosomes (TKDex) (Fig. 8A) [153]. A Schematic illustration of TKDex and cytokine regulation. B Therapeutic regime of RA treatment by TKDex. C The size of spleen after treatment. D, E Represent cytokine level in blood. F Quantification of regulatory T cell after treatment of TKDex [153]. Reproduced with permission, Copyright 2021, Elsevier. G Schematic illustration of neutrophil-derived exosomes (NEs-Exo). H Illustration for therapeutic regime [175]. Reproduced with permission, Copyright 2021, Elsevier They hypothesized that PEG containing ROS-responsive moieties on the exosomal surface protected the exosomes in the bloodstream, while facilitating the exposure of exosomes in the inflamed joint, leading to their uptake into DCs, macrophages, or T cells in the RAM to induce the immunomodulation. The authors first induced immunosuppressive tolerogenic dendritic cells (TolDCs) via preconditioning DCs 2.4 cells with 40 ng/mL IL-10 for 24 h. Subsequently, the dendritic cell-derived exosomes (TolDex) were isolated utilizing TFF systems equipped with an ultrafiltration filter capsule (MWCO = 300 kDa). Meanwhile, the ROS-sensitive linkage (DSPE-TK-mPEG) for exosomal surface modification was synthesized by conjugation of thioketal (TK) linker, methoxy PEG, and 1,2-distearoyl-sn-glycero-phosphoethanolamine (DSPE). Following TolDex isolation, the DSPE-TK-mPEG was modified on the surface of TolDex via the hydrophobic insertion method. The hydrophobic insertion was performed by incubating DMSO-dissolved DSPE-TK-mPEG with TolDex. TKDex was internalized into mature DCs (mDCs) activated with LPS, whereas TolDex modified with PEG without TK (PEGDex) showed insignificant cellular uptake, suggesting its stimuli-sensitive behaviors. The in vitro data exhibited that pro-inflammatory factors were downregulated in mDCs and T cell proliferation was suppressed. In CIA, the negligible difference in the joint-to-liver ratio of TKDex and PEGDex implied that TK moiety did not contribute to former’s degree of accumulation in the lesions. Decrease in IL-6 level, increased TGF-β level, and increased proportion of Tregs suggested that TKDex could modulate T cells as well as DCs in inflamed joints. It is noteworthy that the therapeutic activity of Dex are induced in only ROS-overproduced RAM. Zhang et al. modified ultrasmall Prussian blue nanoparticles (uPB), nanoenzyme, on the surface of neutrophil-derived exosomes to regulate the RAM (Fig. 8G) [175]. Since LFA-1 on the activated neutrophil membrane can bind with activated FLSs, chondrocytes, and macrophages via intracellular activation molecule 1 (ICAM-1), LPS was injected into mice, followed by the isolation of neutrophils. Subsequently, the neutrophil-derived exosomes (NEs-Exo), possessing the LFA-1, were isolated by differential centrifugation. In addition, an antioxidative enzyme mimic, Prussian blue was fabricated into ultrasmall nanoparticles and further anchored to the exosomal surface to relieve the ROS-abundant conditions in inflamed joints. DBCO-sulfo-NHS was conjugated on the exosomal surface in advance, and N3-uPB was linked by copper-free click chemistry. The uPB-Exo showed enhanced uptake into activated FLSs, chondrocytes, and macrophages, as well as ROS scavenging in vitro, indicating the role of LFA-1 in active targeting and activity of nanozymes, respectively. Moreover, uPB-Exo revealed protective effects against oxidative stress and cytokines secreted from apoptotic cells by mimicking SOD-2 and inhibiting apoptosis of activated FLSs, chondrocytes, and macrophages. The authors also validated that apoptosis was related PI3K/AKT signaling pathway. In CIA, both NEs-Exo and uPB-Exo were highly accumulated into paws and penetrated deep into the cartilage. The significant differences in the reduction of TNF-α, IL-1β, and Th1, as well as the increase in Treg count suggested the synergistic effects of uPB with NEs-Exo. M2 macrophage-derived exosomes loaded with a plasmid DNA encoding IL-10 (IL-10 pDNA) and synthetic glucocorticoid were developed for macrophage re-polarization in RA (Fig. 9A) [176]. A Schematic illustration of M2-macrophage derived exosomes (M2-Exo/pDNA). B Therapeutic regime of M2-Exo/pDNA and macrophage polarization. C Photographs of paw thickness [176]. Reproduced with permission, Copyright 2022, Elsevier. D Schematic illustration of anti-inflammatory exosomes (AI-Exo). E Photoacoustic images of joint. F skeletal structure of paw joint [177]. Reproduced with permission, Copyright 2022, Royal Society of Chemistry The authors aimed to inhibit pro-inflammatory factors and promote M1 polarization to M2 macrophages. M2 macrophages (RAW 264.7 cells) were induced with 100 ng/mL IL-4. The pDNA-lipid complexes, comprising earlier synthesized IL-10 pDNA and transfection agent, were incubated with M2 macrophages at 37 ℃ for 3 days. Subsequently, the IL-10 pDNA encapsulated M2-derived exosomes (M2 Exo/pDNA) were isolated using differential centrifugation with ultrafiltration. In brief, supernatant centrifuged at 300 g, 2000 g, and 10,000 g was concentrated using ultrafiltration through MWCO 100 kDa filter, followed by ultracentrifugation at 120,000 g. Lastly, to obtain the betamethasone sodium phosphate (BSP) loaded M2 Exo/pDNA (M2 Exo/pDNA/BSP), the M2 Exo/pDNA and BSP were mixed (wt ratio 1:2) with 80 nM trehalose and electroporated with poring pulse at 200 V, pulsed twice for 5 ms and transfer pulse for five pulses at 20 V for 50 ms at room temperature. M2 Exo/pDNA/BSP showed sustained release of BSP (approximately 98.93% within 20 h). The enhanced uptake of M2 Exo into activated macrophages was attributed to the overexpression of LFA-1 or VLA-4 on M2 Exo, which can bind to ICAM-1 or VCAM-1 on activated macrophages, suggesting the inherent targetability of M2 Exo to M1 macrophages. As expected, the M2 Exo/pDNA/BSP exhibited maximal effects on M1 to M2 polarization compared to free BSP, pDNA/BSP, M2 Exo, M2 Exo/pDNA, and M2 Exo/BSP. In CIA, highly accumulated M2 Exo/pDNA/BSP in inflamed joints regulated pro-and anti-inflammatory cytokines and protected the cartilage, without inducing hepatotoxicity; Thus, this system led to effective co-delivery of genes and drugs without side effects allowing synergy. Tang et al. utilized an external energy source to improve the therapeutic effects in RA (Fig. 9D) [177]. The authors prepared anti-inflammatory exosomes (AI-Exo) by loading IL-10 into macrophage (M0 type)-derived exosomes and irradiated using non-invasive ultrasound (US) during therapy to enhance blood vessel permeability to accelerate drug uptake into tissues and cells. The AI-Exo was prepared as follows; 1) Exosomes were isolated from RAW 264.7 cells using ExoQuick-TC®ULTRA EV Isolation kit, and 2) IL-10 and exosomes were electroporated (wt ratio 2:15, 600 V for 90 μs, 5 times pulses with 1 s interval). The in vitro data showed that US irradiation enhanced the cellular uptake of AI-Exo into activated macrophages. Similarly, the US augmented the accumulation of AI-Exo into inflamed joints in CIA, alleviating RA symptoms. The mechanistic analysis verified that the therapeutic effect of AI-Exo+US group was attributed to the reduction in IL-6 and TNF-α level and decrease CD86/CD206 ratio in macrophages. The authors noted that IL-10 plays a role in promoting polarization of M1 macrophages to M2 macrophages, and US can enhance the targetability by promoting the activated macrophage phagocytosis of AI-Exo. Engineering other subpopulations of extracellular vesicles, including matrix-bound nanovesicles (MBV) and MV, was also reported. The MBVs are similar to MV; however, they are anchored to the extracellular matrix (ECM) and identified as functional components of ECM [206]. Crum et al. verified that the MBV isolated from urinary bladder ECM by enzymatic digestion attenuated the inflammation in pristane-induced arthritis via synovial and splenic macrophage polarization from M1 to M2 phenotype [207]. Meanwhile, MV was used for develo** biomimetic nanoparticles. The zeolitic imidazolate framework-8 (ZIF-8), one of the common metal–organic frameworks (MOF), encapsulated the MTX and was camouflaged with macrophage-derived MV and further modified with DSPE-PEG-FA by hydrophobic insertion to enhance the biocompatibility and targetability [208]. In the same group, the Dex-encapsulated and macrophage-derived MV-coated ZIF-8 was also developed for targeted delivery to arthritic joints [209]. The macrophage-derived MV was employed to coat the poly(lactic-co-glycolic acid) (PLGA) nanoparticles, eliciting enhanced RA targeting [210]. Considering the biological effects of the exosome in cell-to-cell communication, using an exosome as an RA therapeutics is a potential approach for treating RA. In addition to DC and T cells, exosomes derived from various cells, such as NK cells, can enhance each cell's function and modulate the disease microenvironment. Although the etiology of RA has not been precisely elucidated, therapeutic strategies that convert autoimmunity into a state of immune tolerance have been proposed. Because systemic immunosuppression induces undesirable immunodeficiency, the importance of inducing antigen-specific tolerance is emerging to treat autoimmune diseases effectively [211]. Recently, OA or chronic multiple sclerosis was reportedly treated in an antigen-specific manner by inducing tolerogenic DC using nanovaccines utilizing antigen-specificity [212, 213]. Although Rheumavax, an autologous DC modified by exposure to citrullinated peptides, was introduced several years ago and was safe and effective in clinical trials with a single administration, issues related to cell-based therapy remain a challenge. In this perspective, exosomes are non-immunogenic and less toxic than organic/inorganic therapeutic nanoparticles; therefore, they can be used to substitute cell-based therapy. Moreover, DC-derived exosomes can be developed into tolerogenic nanovaccines through various engineering methods. Development of antigen-specific nanovaccines using DC exosomes is a promising strategy for develo** novel RA therapeutics, thereby preventing non-specific immune system downregulation. Challenges such as low in vivo stability and unspecific uptake of bare exosomes have been identified, the need to utilize engineered exosomes has emerged. Engineered exosomes equipped with targeting moieties or PEGylation for long-term circulation have emerged as excellent alternatives as they overcome low in vivo biostability of bare exosomes. However, considering that mass production of engineered exosomes is challenging, the issues related to their producibility and cost need to be addressed. Some strategies, such as three-dimensional cultivation using hallow-fiber bioreactor [214], and focused US irradiation [215], can improve the production efficiency; however, other challenges remain. In addition, the exosomes have a complex structure compared to conventional drugs. Therefore, difficulties in their characterization required for drug approval hinders further application of engineered exosomes in clinical settings. For example, problems of animal serum retention in the cell culture medium used for exosome production can be a huddle for the clinical application of exosomes, as well as the limitation of the human application of stem cells. Exosome-based therapies, encompassing bare and engineered exosomes, offer promising prospects for RA treatment. Even though both strategies have been studied with various types of exosomes, it is crucial to weigh the potential advantages against the limitation of each approach. Bare exosomes possess inherent immunomodulatory properties depending on the origin cell types, and autologous or allogeneic exosomes are biocompatible and have lower immunogenicity, minimizing the risk of immune responses or adverse reactions. Nevertheless, the variability of naturally controlled cargoes might lead to inconsistent therapeutic efficacy. In addition, since the mode of action of bare exosomes' immunomodulatory effects and anti-inflammatory effects are complex and unpredictable, the diverse exosomal content and interaction with various types of cells might lead to undesirable biological effects. On the other hand, various therapeutic cargo like anti-inflammatory drugs, disease-modifying molecules, and immunomodulatory molecules can be loaded within engineered exosomes, and the loading efficiency can be precisely controlled. The targeting moieties on exosomal surfaces enable exosome delivery at the intended site of action, resulting in enhanced therapeutic efficacy and lower side effects. Furthermore, the engineered exosomes can be personalized by customizing to the needs of individual patients based on the heterogeneity of specific diseases. Since the engineered exosomes have more chance to recognize immune systems than bare exosomes, the engineering strategy should augment the biocompatibility for lowing the immunogenicity. In addition to using the exosome directly, in situ exosome biogenesis is an emerging strategy. In situ exosome biogenesis modulation is a strategy without the production process of the exosome, which is expensive. For example, the strategy to inhibit the secretion of exosomes derived from the cell population that overproduces exosomes in the disease microenvironment showed promising microenvironment modulation capability in the pre-clinical state. Conversely, strategies to induce the secretion of exosomes from suppressed cell populations within the disease microenvironment can be considered. Because exosomes function as cell avatars, increased secretion of exosomes can be utilized to restore, reinvigorate, or suppress biological functions within the microenvironment of the suppressor cell population. In conclusion, using exosomes as a potential therapeutic approach for RA is promising due to their non-immunogenicity and less toxicity than other therapeutic nanoparticles. However, issues related to the production and characterization of engineered exosomes need to be addressed. Therefore, further research is required to address these challenges and fully utilize the potential of exosomes in treating RA and other autoimmune diseases. Not applicable. Ascorbic acid N-azidoacetyl-D-mannosamine Antigen-induced arthritis Annexin A1 Antigen-presenting cells Biolayer interferometry Bone marrow-derived macrophages Bovine milk-derived exosomes Betamethasone sodium phosphate Chemotherapy Chlorin e6 Cholesterol Collagen-induced arthritis Clustered regularly interspaced short palindromic repeats Cytotoxic T lymphocyte-associated antigen-4-Ig Curcumin C-X-C motif chemokine ligand 9 Dendritic cells Dendritic cells-derived exosomes 1,1-Dioctadecyl-3,3,3,3-tetramethylindotricarbocyaine iodide Disease-modifying anti-rheumatic drugs Doxorubicin Dextran sulfate 1,2-Distearoyl-sn-glycero-phosphoethanolamine Delayed hypersensitivity Endothelial cells Epigallocatechin gallate Epidermal growth factor receptor Endosomal sorting complex responsible for transport Extracellular vesicles Folic acid Firoblast-like synoviocytes Gene expression omnibus Good manufacturing practices Gingical mesenchymal stem cells Hyaluronic acid Hydroxychloroquine Human leukocyte antigen High-performance liquid chromatography Human umbilical cord mesenchymal stem cells Intracellular activation molecule 1 Indocyanine green Indoleamine-2,3-dioxygenase Nitric oxide synthase Janus kinase Lipopolysaccharide Mitogen-activated protein kinase Molecular beacon Matrix metalloproteinases Mesenchymal stem cells Multivesicular bodies Microvesicles Nuclear factor kappa B Nitric oxide Nonsteroidal anti-inflammatory drugs Polydopamine Photodynamic therapy Poly(ethylene glycol) Positron emission tomography Platelet-derived exosomes Prostaglandin E2 poly(lactic-co-glycolic acid) Polymorphonuclear leukocyte Paclitaxel Rheumatoid arthritis Rheumatoid arthritis microenvironment Red blood cells Ribonucleoprotein Reactive oxygen species Sodium bicarbonate Spinal cord injury Sonodynamic therapy Single guide RNA Scavenger receptor class A Signal transducers and activates transcription 3 Transcription activator-like effector nucleases Tangential flow filtration T-helper cells Thioketal Tumor necrosis factor-α Triphenylphosphonium T regulatory type 1 cells Regulatory T cells Ultrasound Vascular endothelial growth factor Zinc finger nucleases Zinc sulfide Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–79. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017;8(1):15287. Gao M, Gao W, Papadimitriou JM, Zhang C, Gao J, Zheng M. Exosomes—the enigmatic regulators of bone homeostasis. Bone Res. 2018;6(1):36. Desdín-Micó G, Mittelbrunn M. Role of exosomes in the protection of cellular homeostasis. Cell Adhes Migr. 2017;11(2):127–34. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. **a X, Wang Y, Huang Y, Zhang H, Lu H, Zheng JC. Exosomal miRNAs in central nervous system diseases: biomarkers, pathological mediators, protective factors and therapeutic agents. Prog Neurobiol. 2019;183: 101694. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9. Huda MN, Nafiujjaman M, Deaguero IG, Okonkwo J, Hill ML, Kim T, et al. Potential use of exosomes as diagnostic biomarkers and in targeted drug delivery: progress in clinical and preclinical applications. ACS Biomater Sci Eng. 2021;7(6):2106–49. Zhou B, Xu K, Zheng X, Chen T, Wang J, Song Y, et al. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct Target Ther. 2020;5(1):144. Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. 2020;5(1):145. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6. Li S-p, Lin Z-x, Jiang X-y, Yu X-y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol Sin. 2018;39(4):542–51. Chen C, Sun M, Wang J, Su L, Lin J, Yan X. Active cargo loading into extracellular vesicles: Highlights the heterogeneous encapsulation behaviour. J Extracell Vesicles. 2021;10(13): e12163. Xu Z, Zeng S, Gong Z, Yan Y. Exosome-based immunotherapy: a promising approach for cancer treatment. Mol Cancer. 2020;19(1):160. Yang C, Robbins PD. Immunosuppressive exosomes: a new approach for treating arthritis. Int J Rheumatol. 2012;2012: 573528. Liu C, Zhang W, Li Y, Chang J, Tian F, Zhao F, et al. Microfluidic sonication to assemble exosome membrane-coated nanoparticles for immune evasion-mediated targeting. Nano Lett. 2019;19(11):7836–44. Grassi W, De Angelis R, Lamanna G, Cervini C. The clinical features of rheumatoid arthritis. Eur J Radiol. 1998;27:S18–24. Majithia V, Geraci SA. Rheumatoid arthritis: diagnosis and management. Am J Med. 2007;120(11):936–9. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis — immune cell–fibroblast–bone interactions. Nat Rev Rheumatol. 2022;18(7):415–29. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4(1):18001. Zaiss MM, Joyce Wu H-J, Mauro D, Schett G, Ciccia F. The gut–joint axis in rheumatoid arthritis. Nat Rev Rheumatol. 2021;17(4):224–37. Deane KD, Holers VM. Rheumatoid arthritis pathogenesis, prediction, and prevention: an emerging paradigm shift. Arthritis Rheumatol. 2021;73(2):181–93. Choy EHS, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. New Engl J Med. 2001;344(12):907–16. Zhu Y, Zhao T, Liu M, Wang S, Liu S, Yang Y, et al. Rheumatoid arthritis microenvironment insights into treatment effect of nanomaterials. Nano Today. 2022;42: 101358. Farrugia M, Baron B. The role of TNF-α in rheumatoid arthritis: a focus on regulatory T cells. J Clin Transl Res. 2016;2(3):84–90. Schiff MH. Role of interleukin 1 and interleukin 1 receptor antagonist in the mediation of rheumatoid arthritis. Ann Rheum Dis. 2000;59(suppl 1):i103–8. Narazaki M, Tanaka T, Kishimoto T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev Clin Immunol. 2017;13(6):535–51. Oikonomopoulou K, Diamandis EP, Hollenberg MD, Chandran V. Proteinases and their receptors in inflammatory arthritis: an overview. Nat Rev Rheumatol. 2018;14(3):170–80. Scherer HU, van der Woude D, Toes REM. From risk to chronicity: evolution of autoreactive B cell and antibody responses in rheumatoid arthritis. Nat Rev Rheumatol. 2022;18(7):371–83. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55. Wu Z, Ma D, Yang H, Gao J, Zhang G, Xu K, et al. Fibroblast-like synoviocytes in rheumatoid arthritis: Surface markers and phenotypes. Int Immunopharmacol. 2021;93: 107392. Koedderitzsch K, Zezina E, Li L, Herrmann M, Biesemann N. TNF induces glycolytic shift in fibroblast like synoviocytes via GLUT1 and HIF1A. Sci Rep. 2021;11(1):19385. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthrit Res Ther. 2017;19(1):110. Westhoff CS, Freudiger D, Petrow P, Seyfert C, Zacher J, Kriegsmann J, et al. Characterization of collagenase 3 (matrix metalloproteinase 13) messenger RNA expression in the synovial membrane and synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheumatol. 1999;42(7):1517–27. Zhao W, Zhang C, Shi M, Zhang J, Li M, Xue X, et al. The discoidin domain receptor 2/annexin A2/matrix metalloproteinase 13 loop promotes joint destruction in arthritis through promoting migration and invasion of fibroblast-like synoviocytes. Arthritis Rheumatol. 2014;66(9):2355–67. Harada S, Yamamura M, Okamoto H, Morita Y, Kawashima M, Aita T, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheumatol. 1999;42(7):1508–16. Tu J, Huang W, Zhang W, Mei J, Zhu C. Two main cellular components in rheumatoid arthritis: communication between T cells and fibroblast-like Synoviocytes in the joint synovium. Front Immunol. 2022;13: 922111. Brennan FM, Maini RN, Feldmann M. TNF alpha–a pivotal role in rheumatoid arthritis? Br J Rheumatol. 1992;31(5):293–8. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(8):472–85. Lettesjö H, Nordström E, Ström H, Nilsson B, Glinghammar B, Dahlstedt L, et al. Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions. Scand J Immunol. 1998;48(3):286–92. Santos Savio A, Machado Diaz AC, Chico Capote A, Miranda Navarro J, Rodríguez Alvarez Y, Bringas Pérez R, et al. Differential expression of pro-inflammatory cytokines IL-15Ralpha, IL-15, IL-6 and TNFalpha in synovial fluid from rheumatoid arthritis patients. BMC Musculoskelet Disord. 2015;16:51. Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Rheumatol. 2000;2(3):189–202. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6(1):15. Kim SJ, Shin HH, Park SY, Lee DS, Lee EA, Cho SD, et al. Induction of MMP-13 expression by soluble human glucocorticoid-induced tumor necrosis factor receptor in fibroblast-like synovial cells. Osteoarthr Cartil. 2006;14(2):146–53. Cope AP, Schulze-Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(5):S4. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–82. Roberts CA, Dickinson AK, Taams LS. The interplay between monocytes/Macrophages and CD4+ T cell subsets in rheumatoid arthritis. Front Immunol. 2015;6:571. Esensten JH, Wofsy D, Bluestone JA. Regulatory T cells as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):560–5. Leipe J, Skapenko A, Lipsky PE, Schulze-Koops H. Regulatory T cells in rheumatoid arthritis. Arthrit Res Ther. 2005;7(3):93. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J Exp Med. 2004;200(3):277–85. Möttönen M, Heikkinen J, Mustonen L, Isomäki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. 2005;140(2):360–7. Khan S, Greenberg JD, Bhardwaj N. Dendritic cells as targets for therapy in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):566–71. Wehr P, Purvis H, Law SC, Thomas R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin Exp Immunol. 2019;196(1):12–27. Cifuentes-Rius A, Desai A, Yuen D, Johnston APR, Voelcker NH. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat Nanotechnol. 2021;16(1):37–46. Sarkar S, Fox DA. Dendritic cells in rheumatoid arthritis. Front Biosci. 2005;10:656–65. Wu F, Gao J, Kang J, Wang X, Niu Q, Liu J, et al. B cells in rheumatoid arthritis: pathogenic mechanisms and treatment prospects. Front Immunol. 2021;12: 750753. Yanaba K, Bouaziz J-D, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28(5):639–50. O’Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis1. J Immunol. 2005;174(6):3781–8. Weyand CM, Kurtin PJ, Goronzy JJ. Ectopic lymphoid organogenesis: a fast track for autoimmunity. Am J Clin Pathol. 2001;159(3):787–93. McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat Med. 1997;3(2):189–95. Beech JT, Andreakos E, Ciesielski CJ, Green P, Foxwell BMJ, Brennan FM. T-cell contact-dependent regulation of CC and CXC chemokine production in monocytes through differential involvement of NFκB: implications for rheumatoid arthritis. Arthrit Res Ther. 2006;8(6):R168. Burger D, Dayer JM. The role of human T-lymphocyte-monocyte contact in inflammation and tissue destruction. Arthritis Rheumatol. 2002;4(3):S169-76. Chen J, Li P, Zhang T, Xu Z, Huang X, Wang R, et al. Review on Strategies and technologies for exosome isolation and purification. Front Bioeng Biotechnol. 2021;9: 811971. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83. Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9(1):19. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244–7. Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic. 2009;10(7):925–37. Géminard C, De Gassart A, Blanc L, Vidal M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic. 2004;5(3):181–93. Hurley JH, Odorizzi G. Get on the exosome bus with ALIX. Nat Cell Biol. 2012;14(7):654–5. Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, López-Martín S, Ursa A, Sánchez-Madrid F, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem. 2013;288(17):11649–61. Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6(10):801–11. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30. Song L, Tang S, Han X, Jiang Z, Dong L, Liu C, et al. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat Commun. 2019;10(1):1639. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977. EL Andaloussi S, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–57. Bang C, Thum T. Exosomes: new players in cell–cell communication. Int J Biochem Cell Biol. 2012;44(11):2060–4. Morishita M, Takahashi Y, Nishikawa M, Takakura Y. Pharmacokinetics of exosomes—an important factor for elucidating the biological roles of exosomes and for the development of exosome-based therapeutics. J Pharm Sci. 2017;106(9):2265–9. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–5. Qin J, Xu Q. Functions and application of exosomes. Acta Pol Pharm. 2014;71(4):537–43. Kim S-H, Lechman ER, Bianco N, Menon R, Keravala A, Nash J, et al. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J Immunol. 2005;174(10):6440–8. Withrow J, Murphy C, Liu Y, Hunter M, Fulzele S, Hamrick MW. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthrit Res Ther. 2016;18(1):286. Kato T, Miyaki S, Ishitobi H, Nakamura Y, Nakasa T, Lotz MK, et al. Exosomes from IL-1β stimulated synovial fibroblasts induce osteoarthritic changes in articular chondrocytes. Arthrit Res Ther. 2014;16(4):1–11. Wang Y, Zhao M, Liu S, Guo J, Lu Y, Cheng J, et al. Macrophage-derived extracellular vesicles: diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. 2020;11(10):924. Boriachek K, Islam MN, Möller A, Salomon C, Nguyen N-T, Hossain MSA, et al. Biological functions and current advances in isolation and detection strategies for exosome nanovesicles. Small. 2018;14(6):1702153. Buzas EI, György B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. 2014;10(6):356–64. Lai JJ, Chau ZL, Chen S-Y, Hill JJ, Korpany KV, Liang N-W, et al. Exosome processing and characterization approaches for research and technology development. Adv Sci. 2022;9(15):2103222. Tran T-H, Mattheolabakis G, Aldawsari H, Amiji M. Exosomes as nanocarriers for immunotherapy of cancer and inflammatory diseases. Clin Immunol. 2015;160(1):46–58. Lai RC, Chen TS, Lim SK. Mesenchymal stem cell exosome: a novel stem cell-based therapy for cardiovascular disease. Regen Med. 2011;6(4):481–92. Newton WC, Kim JW, Luo JZ, Luo L. Stem cell-derived exosomes: a novel vector for tissue repair and diabetic therapy. J Mol Endocrinol. 2017;59(4):R155–65. Wang J, Chen P, Dong Y, **e H, Wang Y, Soto F, et al. Designer exosomes enabling tumor targeted efficient chemo/gene/photothermal therapy. Biomaterials. 2021;276: 121056. Yao X, Lyu P, Yoo K, Yadav MK, Singh R, Atala A, et al. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J Extracell Vesicles. 2021;10(5): e12076. Yong T, Zhang X, Bie N, Zhang H, Zhang X, Li F, et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat Commun. 2019;10(1):1–16. Song S, Shim MK, Lim S, Moon Y, Yang S, Kim J, et al. In situ one-step fluorescence labeling strategy of exosomes via bioorthogonal click chemistry for real-time exosome tracking in vitro and in vivo. Bioconj Chem. 2020;31(5):1562–74. Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. OncoImmunology. 2016;5(4): e1071008. Pusic AD, Pusic KM, Clayton BLL, Kraig RP. IFNγ-stimulated dendritic cell exosomes as a potential therapeutic for remyelination. J Neuroimmunol. 2014;266(1):12–23. Pusic KM, Won L, Kraig RP, Pusic AD. IFNγ-Stimulated dendritic cell exosomes for treatment of migraine modeled using spreading depression. Front Neurosci. 2019;13:942. Jia Z, Liu J, Li B, Yi L, Wu Y, **ng J, et al. Exosomes with FOXP3 from gene-modified dendritic cells ameliorate the development of EAE by regulating the balance of Th/Treg. Int J Med Sci. 2022;19(8):1265–74. Wang L, Yu Z, Wan S, Wu F, Chen W, Zhang B, et al. Exosomes derived from dendritic cells treated with Schistosoma japonicum soluble egg antigen attenuate DSS-induced colitis. Front Pharmacol. 2017;8:651. Yang X, Meng S, Jiang H, Chen T, Wu W. Exosomes derived from interleukin-10-treated dendritic cells can inhibit trinitrobenzene sulfonic acid-induced rat colitis. Scand J Gastroenterol. 2010;45(10):1168–77. Cai Z, Zhang W, Yang F, Yu L, Yu Z, Pan J, et al. Immunosuppressive exosomes from TGF-β1 gene-modified dendritic cells attenuate Th17-mediated inflammatory autoimmune disease by inducing regulatory T cells. Cell Res. 2012;22(3):607–10. Rao Q, Ma G, Li M, Wu H, Zhang Y, Zhang C, et al. Targeted delivery of triptolide by dendritic cell-derived exosomes for colitis and rheumatoid arthritis therapy in murine models. Br J Pharmacol. 2023;180(3):330–46. Kim SH, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, et al. Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive. Mol Ther. 2006;13(2):289–300. Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol. 2007;179(4):2242–9. Bianco NR, Kim SH, Ruffner MA, Robbins PD. Therapeutic effect of exosomes from indoleamine 2, 3-dioxygenase–positive dendritic cells in collagen-induced arthritis and delayed-type hypersensitivity disease models. Arthr Rheuma. 2009;60(2):380–9. Kim H, Park H-J, Chang HW, Back JH, Lee SJ, Park YE, et al. Exosome-guided direct reprogramming of tumor-associated macrophages from protumorigenic to antitumorigenic to fight cancer. Bioact Mater. 2022;25:527. Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther. 2017;25(7):1665–75. Liu P, **ong Y, Chen L, Lin C, Yang Y, Lin Z, et al. Angiogenesis-based diabetic skin reconstruction through multifunctional hydrogel with sustained releasing of M2 Macrophage-derived exosome. Chem Eng J. 2022;431: 132413. Kim H, Wang SY, Kwak G, Yang Y, Kwon IC, Kim SH. Exosome-guided phenotypic switch of M1 to M2 macrophages for cutaneous wound healing. Adv Sci. 2019;6(20):1900513. Chen X, Wan Z, Yang L, Song S, Fu Z, Tang K, et al. Exosomes derived from reparative M2-like macrophages prevent bone loss in murine periodontitis models via IL-10 mRNA. J Nanobiotechnology. 2022;20(1):110. Huang Y, Liu Q, Liu L, Huo F, Guo S, Tian W. Lipopolysaccharide-preconditioned dental follicle stem cells derived small extracellular vesicles treating periodontitis via reactive oxygen species/Mitogen-activated protein kinase signaling-mediated antioxidant effect. Int J Nanomed. 2022;17:799. Watanabe Y, Fukuda T, Hayashi C, Nakao Y, Toyoda M, Kawakami K, et al. Extracellular vesicles derived from GMSCs stimulated with TNF-α and IFN-α promote M2 macrophage polarization via enhanced CD73 and CD5L expression. Sci Rep. 2022;12(1):1–16. Tian J, Chen W, **ong Y, Li Q, Kong S, Li M, et al. Small extracellular vesicles derived from hypoxic preconditioned dental pulp stem cells ameliorate inflammatory osteolysis by modulating macrophage polarization and osteoclastogenesis. Bioact Mater. 2023;22:326–42. Zhu F, Wei C, Wu H, Shuai B, Yu T, Gao F, et al. Hypoxic mesenchymal stem cell-derived exosomes alleviate ulcerative colitis injury by limiting intestinal epithelial cells reactive oxygen species accumulation and DNA damage through HIF-1α. Int Immunopharmacol. 2022;113: 109426. Hazrati A, Malekpour K, Soudi S, Hashemi SM. CRISPR/Cas9-engineered mesenchymal stromal/stem cells and their extracellular vesicles: a new approach to overcoming cell therapy limitations. Biomed Pharmacother. 2022;156: 113943. Bobis-Wozowicz S, Kania K, Nit K, Blazowska N, Kmiotek-Wasylewska K, Paw M, et al. Efficient in vivo genome editing mediated by stem-cells derived extracellular vesicles carrying designer nucleases. 2021. PREPRINT available at https://doi.org/10.1101/2021.02.25.432823. Yoon HY, Koo H, Kim K, Kwon IC. Molecular imaging based on metabolic glycoengineering and bioorthogonal click chemistry. Biomaterials. 2017;132:28–36. Lim S, Yoon HY, Park S-J, Song S, Shim MK, Yang S, et al. Predicting in vivo therapeutic efficacy of bioorthogonally labeled endothelial progenitor cells in hind limb ischemia models via non-invasive fluorescence molecular tomography. Biomaterials. 2021;266: 120472. Kim W, Yoon HY, Lim S, Stayton PS, Kim I-S, Kim K, et al. In vivo tracking of bioorthogonally labeled T-cells for predicting therapeutic efficacy of adoptive T-cell therapy. J Contr Rel. 2021;329:223–36. Lim S, Yoon HY, Jang HJ, Song S, Kim W, Park J, et al. Dual-Modal imaging-guided precise tracking of Bioorthogonally labeled mesenchymal stem cells in mouse brain stroke. ACS Nano. 2019;13(10):10991–1007. Yoon HY, Shin ML, Shim MK, Lee S, Na JH, Koo H, et al. Artificial chemical reporter targeting strategy using bioorthogonal click reaction for improving active-targeting efficiency of tumor. Mol Pharm. 2017;14(5):1558–70. Lim GT, You DG, Han HS, Lee H, Shin S, Oh BH, et al. Bioorthogonally surface-edited extracellular vesicles based on metabolic glycoengineering for CD44-mediated targeting of inflammatory diseases. J Extracell Vesicles. 2021;10(5): e12077. You DG, Lim GT, Kwon S, Um W, Oh BH, Song SH, et al. Metabolically engineered stem cell–derived exosomes to regulate macrophage heterogeneity in rheumatoid arthritis. Sci Adv. 2021;7(23):eabe0083. Wang J, Li W, Zhang L, Ban L, Chen P, Du W, et al. Chemically edited exosomes with dual ligand purified by microfluidic device for active targeted drug delivery to tumor cells. ACS Appl Mater Interfaces. 2017;9(33):27441–52. Chen G, Zhu JY, Zhang ZL, Zhang W, Ren JG, Wu M, et al. Transformation of cell-derived microparticles into quantum-dot-labeled nanovectors for antitumor siRNA delivery. Angew Chem. 2015;127(3):1050–4. Sancho-Albero M, del Mar E-B, Beltran-Visiedo M, Fernandez-Messina L, Sebastian V, Sanchez-Madrid F, et al. Efficient encapsulation of theranostic nanoparticles in cell-derived exosomes: leveraging the exosomal biogenesis pathway to obtain hollow gold nanoparticle-hybrids. Nanoscale. 2019;11(40):18825–36. Sancho-Albero M, Navascués N, Mendoza G, Sebastián V, Arruebo M, Martín-Duque P, et al. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J Nanobiotechnology. 2019;17(1):1–13. Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. 2021;16(7):748–59. Chen S, Sun F, Qian H, Xu W, Jiang J. Preconditioning and engineering strategies for improving the efficacy of mesenchymal stem cell-derived exosomes in cell-free therapy. Stem Cells Int. 2022;2022:1779346. Zhang M, Shao W, Yang T, Liu H, Guo S, Zhao D, et al. Conscription of immune cells by light-activatable silencing NK-derived exosome (LASNEO) for synergetic tumor eradication. Adv Sci. 2022;9(22):2201135. Han Z, Lv W, Li Y, Chang J, Zhang W, Liu C, et al. Improving tumor targeting of exosomal membrane-coated polymeric nanoparticles by conjugation with aptamers. ACS Appl Bio Mater. 2020;3(5):2666–73. Pham TC, Jayasinghe MK, Pham TT, Yang Y, Wei L, Usman WM, et al. Covalent conjugation of extracellular vesicles with peptides and nanobodies for targeted therapeutic delivery. J Extracell Vesicles. 2021;10(4): e12057. Ma F, Feng J, Liu X, Tian Y, Wang W-J, Luan F-X, et al. A synergistic therapeutic nano-eyedrop for dry eye disease based on ascorbic acid-coupled exosomes. Nanoscale. 2023;15(4):1890–9. Wan T, Zhong J, Pan Q, Zhou T, ** Y, Liu X. Exosome-mediated delivery of Cas9 ribonucleoprotein complexes for tissue-specific gene therapy of liver diseases. Sci Adv. 2022;8(37):eabp9435. Almeida SFF, Fonseca A, Sereno J, Ferreira HRS, Lapo-Pais M, Martins-Marques T, et al. Osteosarcoma-derived exosomes as potential PET imaging nanocarriers for lung metastasis. Small. 2022;18(49):2203999. Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Contr Rel. 2015;219:396–405. Fu S, Wang Y, **a X, Zheng JC. Exosome engineering: current progress in cargo loading and targeted delivery. NanoImpact. 2020;20: 100261. Kwon S, Shin S, Do M, Oh BH, Song Y, Bui VD, et al. Engineering approaches for effective therapeutic applications based on extracellular vesicles. J Contr Rel. 2021;330:15–30. Cui J, Wang X, Li J, Zhu A, Du Y, Zeng W, et al. Immune exosomes loading self-assembled nanomicelles traverse the blood-brain barrier for chemo-immunotherapy against glioblastoma. ACS Nano. 2023;17(2):1464–84. Nguyen Cao TG, Kang JH, You JY, Kang HC, Rhee WJ, Ko YT, et al. Safe and targeted sonodynamic cancer therapy using biocompatible exosome-based nanosonosensitizers. ACS Appl Mater Interfaces. 2021;13(22):25575–88. Nguyen Cao TG, Kang JH, Kim W, Lim J, Kang SJ, You JY, et al. Engineered extracellular vesicle-based sonotheranostics for dual stimuli-sensitive drug release and photoacoustic imaging-guided chemo-sonodynamic cancer therapy. Theranostics. 2022;12(3):1247–66. Nguyen Cao TG, Truong Hoang Q, Hong EJ, Kang SJ, Kang JH, Ravichandran V, et al. Mitochondria-targeting sonosensitizer-loaded extracellular vesicles for chemo-sonodynamic therapy. J Contr Rel. 2023;354:651–63. Liu W, Wei L, Li M, Mo J. Zinc sulfide-based hybrid exosome-coated nanoplatform for targeted treatment of glioblastoma in an orthotopic mouse glioblastoma model. Mater Today Adv. 2023;17: 100327. Wang B, Chang M, Zhang R, Wo J, Wu B, Zhang H, et al. Spinal cord injury target-immunotherapy with TNF-α autoregulated and feedback-controlled human umbilical cord mesenchymal stem cell derived exosomes remodelled by CRISPR/Cas9 plasmid. Biomat Adv. 2022;133: 112624. McAndrews KM, **ao F, Chronopoulos A, LeBleu VS, Kugeratski FG, Kalluri R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras<sup>G12D</sup> in pancreatic cancer. Life Sci Alliance. 2021;4(9): e202000875. Hade MD, Suire CN, Suo Z. An effective peptide-based platform for efficient exosomal loading and cellular delivery of a microRNA. ACS Appl Mater Interfaces. 2023;15(3):3851–66. Ranjan P, Colin K, Dutta RK, Verma SK. Challenges and future scope of exosomes in the treatment of cardiovascular diseases. J Physiol. 2022. https://doi.org/10.1113/JP282053. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Del Rev. 2016;99:28–51. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503. Cheng L, Zhang X, Tang J, Lv Q, Liu J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials. 2021;275: 120964. Belhadj Z, He B, Deng H, Song S, Zhang H, Wang X, et al. A combined “eat me/don’t eat me” strategy based on extracellular vesicles for anticancer nanomedicine. J Extracell Vesicles. 2020;9(1):1806444. Huang H, Yi X, Wei Q, Li M, Cai X, Lv Y, et al. Edible and cation-free kiwi fruit derived vesicles mediated EGFR-targeted siRNA delivery to inhibit multidrug resistant lung cancer. J Nanobiotechnol. 2023;21(1):41. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mat. 2013;12(11):991–1003. Lee ES, Sul JH, Shin JM, Shin S, Lee JA, Kim HK, et al. Reactive oxygen species-responsive dendritic cell-derived exosomes for rheumatoid arthritis. Acta Biomater. 2021;128:462–73. Hock ES, Martyn-St James M, Wailoo A, Scott DL, Stevenson M, Rawdin A, et al. Treat-to-target strategies in rheumatoid arthritis: a systematic review and cost-effectiveness analysis. SN Compre Clin Med. 2021;3(3):838–54. Wu H, Zhou X, Wang X, Cheng W, Hu X, Wang Y, et al. miR-34a in extracellular vesicles from bone marrow mesenchymal stem cells reduces rheumatoid arthritis inflammation via the cyclin I/ATM/ATR/p53 axis. J Cell Mol Med. 2021;25(4):1896–910. Meng Q, Qiu B. Exosomal microRNA-320a derived from mesenchymal stem cells regulates rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing CXCL9 expression. Front Physiol. 2020;11:441. Tian X, Wei W, Cao Y, Ao T, Huang F, Javed R, et al. Gingival mesenchymal stem cell-derived exosomes are immunosuppressive in preventing collagen-induced arthritis. J Cell Mol Med. 2022;26(3):693–708. Ma D, Xu K, Zhang G, Liu Y, Gao J, Tian M, et al. Immunomodulatory effect of human umbilical cord mesenchymal stem cells on T lymphocytes in rheumatoid arthritis. Int Immunopharm. 2019;74: 105687. Xu K, Ma D, Zhang G, Gao J, Su Y, Liu S, et al. Human umbilical cord mesenchymal stem cell-derived small extracellular vesicles ameliorate collagen-induced arthritis via immunomodulatory T lymphocytes. Mol Immunol. 2021;135:36–44. Arntz OJ, Pieters BCH, Oliveira MC, Broeren MGA, Bennink MB, de Vries M, et al. Oral administration of bovine milk derived extracellular vesicles attenuates arthritis in two mouse models. Mol Nutr Food Res. 2015;59(9):1701–12. Zhu D, Tian J, Wu X, Li M, Tang X, Rui K, et al. G-MDSC-derived exosomes attenuate collagen-induced arthritis by impairing Th1 and Th17 cell responses. Biochim Biophys Acta - Mol Basis Dis. 2019;1865(12): 165540. Cosenza S, Toupet K, Maumus M, Luz-Crawford P, Blanc-Brude O, Jorgensen C, et al. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics. 2018;8(5):1399–410. Wu X, Zhu D, Tian J, Tang X, Guo H, Ma J, et al. Granulocytic myeloid-derived suppressor cell exosomal prostaglandin E2 ameliorates collagen-induced arthritis by enhancing IL-10+ B cells. Front Immunol. 2020;11: 588500. Meng H-Y, Chen L-Q, Chen L-H. The inhibition by human MSCs-derived miRNA-124a overexpression exosomes in the proliferation and migration of rheumatoid arthritis-related fibroblast-like synoviocyte cell. BMC Musculoskel Disord. 2020;21(1):150. Zheng J, Zhu L, Iok In I, Chen Y, Jia N, Zhu W. Bone marrow-derived mesenchymal stem cells-secreted exosomal microRNA-192-5p delays inflammatory response in rheumatoid arthritis. Int Immunopharmacol. 2020;78: 105985. Chen Z, Wang H, **a Y, Yan F, Lu Y. Therapeutic potential of mesenchymal cell-derived miRNA-150-5p–expressing exosomes in rheumatoid arthritis mediated by the modulation of MMP14 and VEGF. J Immunol. 2018;201(8):2472–82. Takenaka M, Yabuta A, Takahashi Y, Takakura Y. Interleukin-4-carrying small extracellular vesicles with a high potential as anti-inflammatory therapeutics based on modulation of macrophage function. Biomaterials. 2021;278: 121160. Kim H, Back JH, Han G, Lee SJ, Park YE, Gu MB, et al. Extracellular vesicle-guided in situ reprogramming of synovial macrophages for the treatment of rheumatoid arthritis. Biomaterials. 2022;286: 121578. Kay AG, Treadwell K, Roach P, Morgan R, Lodge R, Hyland M, et al. Therapeutic effects of hypoxic and pro-inflammatory priming of mesenchymal stem cell-derived extracellular vesicles in inflammatory arthritis. Int J Mol Sci. 2022;23(1):126. Top** LM, Thomas BL, Rhys HI, Tremoleda JL, Foster M, Seed M, et al. Targeting extracellular vesicles to the arthritic joint using a damaged cartilage-specific antibody. Front Immunol. 2020;11:10. Song C, Xu S, Chang L, Zhao X, Mei X, Ren X, et al. Preparation of EGCG decorated, injectable extracellular vesicles for cartilage repair in rat arthritis. Regen Biomater. 2021;8(6):067. Ma Q, Bai J, Xu J, Dai H, Fan Q, Fei Z, et al. Resha** the inflammatory environment in rheumatoid arthritis joints by targeting delivery of berberine with platelet-derived extracellular vesicles. Adv NanoBiomed Res. 2021;1(11):2100071. He X, Zhang C, Amirsaadat S, Jalil AT, Kadhim MM, Abasi M, et al. Curcumin-loaded mesenchymal stem cell-derived exosomes efficiently attenuate proliferation and inflammatory response in rheumatoid arthritis fibroblast-like Synoviocytes. Appl Biochem Biotechnol. 2022;195:51. Yan F, Zhong Z, Wang Y, Feng Y, Mei Z, Li H, et al. Exosome-based biomimetic nanoparticles targeted to inflamed joints for enhanced treatment of rheumatoid arthritis. J Nanobiotechnol. 2020;18(1):1–15. Zhang L, Qin Z, Sun H, Chen X, Dong J, Shen S, et al. Nanoenzyme engineered neutrophil-derived exosomes attenuate joint injury in advanced rheumatoid arthritis via regulating inflammatory environment. Bioact Mater. 2022;18:1–14. Li H, Feng Y, Zheng X, Jia M, Mei Z, Wang Y, et al. M2-type exosomes nanoparticles for rheumatoid arthritis therapy via macrophage re-polarization. J Contr Rel. 2022;341:16–30. Tang Y, Wu Z, Guo R, Huang J, Rong X, Zhu B, et al. Ultrasound-augmented anti-inflammatory exosomes for targeted therapy in rheumatoid arthritis. J Mater Chem B. 2022;10:7862. Li Z, Yuan Y, Zhang Z, Zhang X, Yang H, Li H, et al. Cell penetrating peptide modified M2 macrophage derived exosomes treat spinal cord injury and rheumatoid arthritis by loading curcumin. Mater Des. 2023;225: 111455. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–33. Németh T, Nagy G, Pap T. Synovial fibroblasts as potential drug targets in rheumatoid arthritis, where do we stand and where shall we go? Ann Rheum Dis. 2022;81(8):1055–64. Wang W, Deng Z, Liu G, Yang J, Zhou W, Zhang C, et al. Platelet-derived extracellular vesicles promote the migration and invasion of rheumatoid arthritis fibroblast-like synoviocytes via CXCR2 signaling. Exp Ther Med. 2021;22(4):1120. Chen Y, Dang J, Lin X, Wang M, Liu Y, Chen J, et al. RA fibroblast-like synoviocytes derived extracellular vesicles promote angiogenesis by miRNA-1972 targeting p53/mTOR signaling in vascular endotheliocyte. Front Immunol. 2022;13:793855. Mellado M, Martínez-Muñoz L, Cascio G, Lucas P, Pablos JL, Rodríguez-Frade JM. T cell migration in rheumatoid arthritis. Front Immunol. 2015;6:384. Cope AP. T cells in rheumatoid arthritis. Arthrit Res Ther. 2008;10(1):S1. Zhang Y, Wang Z, Shi B, Li Y, Wang R, Sun J, et al. Effect of gingival mesenchymal stem cell-derived exosomes on inflammatory macrophages in a high-lipid microenvironment. Int Immunopharm. 2021;94: 107455. Wang R, Ji Q, Meng C, Liu H, Fan C, Lipkind S, et al. Role of gingival mesenchymal stem cell exosomes in macrophage polarization under inflammatory conditions. Int Immunopharm. 2020;81: 106030. Chatterton DEW, Nguyen DN, Bering SB, Sangild PT. Anti-inflammatory mechanisms of bioactive milk proteins in the intestine of newborns. Int J Biochem Cell Biol. 2013;45(8):1730–47. Han G, Cho H, Kim H, Jang Y, Jang H, Kim DE, et al. Bovine colostrum derived-exosomes prevent dextran sulfate sodium-induced intestinal colitis via suppression of inflammation and oxidative stress. Biomater Sci. 2022;10(8):2076–87. Feng X, Chen X, Zheng X, Zhu H, Qi Q, Liu S, et al. Latest trend of milk derived exosomes: cargos, functions, and applications. Front nutr. 2021;8:747294. Schulze-Koops H, Kalden JR. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2001;15(5):677–91. Ascanius SR, Hansen MS, Ostenfeld MS, Rasmussen JT. Milk-derived extracellular vesicles suppress inflammatory cytokine expression and nuclear factor-κB activation in lipopolysaccharide-stimulated macrophages. Dairy. 2021;2(2):165–78. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther. 2003;5(4):S1. Bugatti S, Vitolo B, Caporali R, Montecucco C, Manzo A. B cells in rheumatoid arthritis: from pathogenic players to disease biomarkers. Biomed Res Int. 2014;2014: 681678. Millan CLB, Weeratna R, Krieg AM, Siegrist C-A, Davis HL. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc Natl Acad Sci USA. 1998;95(26):15553–8. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–93. Kim H-J, Berek C. B cells in rheumatoid arthritis. Arthrit Res Ther. 2000;2(2):126. Kim H-J, Krenn V, Steinhauser G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. 1999;162(5):3053–62. Yu M, Cavero V, Lu Q, Li H. Follicular helper T cells in rheumatoid arthritis. Clin Rheumatol. 2015;34(9):1489–93. Rhys HI, Dell’Accio F, Pitzalis C, Moore A, Norling LV, Perretti M. Neutrophil microvesicles from healthy control and rheumatoid arthritis patients prevent the inflammatory activation of macrophages. EBioMedicine. 2018;29:60–9. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med. 2015;7(315):315ra190-315ra190. Lo Sicco C, Reverberi D, Balbi C, Ulivi V, Principi E, Pascucci L, et al. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects: endorsement of macrophage polarization. Stem Cells Transl Med. 2017;6(3):1018–28. Donate PB, Alves de Lima K, Peres RS, Almeida F, Fukada SY, Silva TA, et al. Cigarette smoke induces miR-132 in Th17 cells that enhance osteoclastogenesis in inflammatory arthritis. Proc Natl Acad Sci USA. 2021;118(1):e2017120118. Yoon HY, Lee D, Lim D-K, Koo H, Kim K. Copper-free click chemistry: applications in drug delivery, cell tracking, and tissue engineering. Adv Mat. 2022;34(10):2107192. Takayama Y, Kusamori K, Nishikawa M. Click chemistry as a tool for cell engineering and drug delivery. Molecules. 2019;24(1):172. Huleihel L, Hussey GS, Naranjo JD, Zhang L, Dziki JL, Turner NJ, et al. Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv. 2016;2(6): e1600502. Crum RJ, Hall K, Molina CP, Hussey GS, Graham E, Li H, et al. Immunomodulatory matrix-bound nanovesicles mitigate acute and chronic pristane-induced rheumatoid arthritis. NPJ Regen Med. 2022;7(1):13. Wang Y, Jia M, Zheng X, Wang C, Zhou Y, Pan H, et al. Microvesicle-camouflaged biomimetic nanoparticles encapsulating a metal-organic framework for targeted rheumatoid arthritis therapy. J Nanobiotechnology. 2022;20(1):253. Wang Y, Zhang D, Jia M, Zheng X, Liu Y, Wang C, et al. ZIF-8 nanoparticles coated with macrophage-derived microvesicles for sustained, targeted delivery of dexamethasone to arthritic joints. J Drug Target. 2022;30:1–11. Li R, He Y, Zhu Y, Jiang L, Zhang S, Qin J, et al. Route to rheumatoid arthritis by macrophage-derived microvesicle-coated nanoparticles. Nano Lett. 2019;19(1):124–34. Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7(9):665–77. Sohn HS, Choi JW, Jhun J, Kwon SP, Jung M, Yong S, et al. Tolerogenic nanoparticles induce type II collagen–specific regulatory T cells and ameliorate osteoarthritis. Sci Adv. 2022;8(47):eabo5284. Nguyen TL, Choi Y, Im J, Shin H, Phan NM, Kim MK, et al. Immunosuppressive biomaterial-based therapeutic vaccine to treat multiple sclerosis via re-establishing immune tolerance. Nat Commun. 2022;13(1):1–15. Yan L, Wu X. Exosomes produced from 3D cultures of umbilical cord mesenchymal stem cells in a hollow-fiber bioreactor show improved osteochondral regeneration activity. Cell Biol Toxicol. 2020;36:165–78. Sheybani ND, Batts AJ, Mathew AS, Thim EA, Price RJ. Focused ultrasound hyperthermia augments release of glioma-derived extracellular vesicles with differential immunomodulatory capacity. Theranostics. 2020;10(16):7436. Not applicable. This work was supported by the DGIST R&D Program of the Ministry of Science and ICT, grant number 2020010096. It was also supported by the DGIST Start-up Fund Program of the Ministry of Science and ICT (2022030138). Conceptualization, E.S.L. and J.M.S.; Investigation, E.S.L., H.K., C.H.K., and J.M.S.; Manuscript Preparation, E.S.L., C.H.K., and J.M.S.; Visualization and Editing, E.S.L., C.H.K., and J.M.S.; Reviewing, H.-C.K., S.-K.C., S.W.J., S.-G.L., S.-J.L., H.-K.N., J.H.P.; Supervision, J.M.S.. All authors have read and approved the manuscript for publication. Not applicable. Not applicable. Not applicable. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data. Lee, E.S., Ko, H., Kim, C.H. et al. Disease-microenvironment modulation by bare- or engineered-exosome for rheumatoid arthritis treatment.
Biomater Res 27, 81 (2023). https://doi.org/10.1186/s40824-023-00418-2 Received: Accepted: Published: DOI: https://doi.org/10.1186/s40824-023-00418-2Rheumatoid arthritis

Exosome
Biogenesis of exosomes
Engineering

Pre-isolation modification


Others


Challenges and perspectives
Conclusion
Availability of data and materials
Abbreviations
References
Acknowledgements
Funding
Author information
Authors and Affiliations
Contributions
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Additional information
Publisher’s Note
Rights and permissions
About this article
Cite this article
Keywords
