
Abstract
Background
The etiology of nonalcoholic fatty liver disease (NAFLD) involves a complex interaction of genetic and environmental factors. Previous observational studies have revealed that higher leptin levels are related to a lower risk of develo** NAFLD, but the causative association remains unknown. We intended to study the causal effect between leptin and NAFLD using the Mendelian randomization (MR) study.
Methods
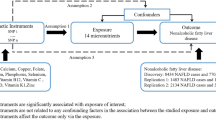
We performed a two-sample Mendelian randomization (TSMR) analysis using summary GWAS data from leptin (up to 50,321 individuals) and NAFLD (8,434 cases and 770,180 controls) in a European population. Instrumental variables (IVs) that satisfied the three core assumptions of Mendelian randomization were selected. The TSMR analysis was conducted using the inverse variance weighted (IVW) method, MR-Egger regression method, and weighted median (WM) method. To ensure the accuracy and stability of the study results, heterogeneity tests, multiple validity tests, and sensitivity analyses were conducted.
Results
The findings of the TSMR correlation analysis between NAFLD and leptin were as follows: IVW method (odds ratio (OR) 0.6729; 95% confidence interval (95% CI) 0.4907–0.9235; P = 0.0142), WM method (OR 0.6549; 95% CI 0.4373–0.9806; P = 0.0399), and MR-Egger regression method (P = 0.6920). Additionally, the findings of the TSMR correlation analysis between NAFLD and circulating leptin levels adjusted for body mass index (BMI) were as follows: IVW method (OR 0.5876; 95% CI 0.3781–0.9134; P = 0.0181), WM method (OR 0.6074; 95% CI 0.4231–0.8721; P = 0.0069), and MR-Egger regression method (P = 0.8870). It has also been shown that higher levels of leptin are causally linked to a lower risk of develo** NAFLD, suggesting that leptin may serve as a protective factor for NAFLD.
Conclusions
Using TSMR analysis and the GWAS database, we investigated the genetic relationship between elevated leptin levels and lowered risk of NAFLD in this study. However, further research is required to understand the underlying mechanisms.
Similar content being viewed by others


Background
Over the past two decades, nonalcoholic fatty liver disease (NAFLD) has progressed from a relatively unknown disease to the leading cause of chronic liver disease worldwide [1]. Its global frequency is quickly increasing, reaching up to 25% in developed countries like the United States [2]. NAFLD is a degenerative disease caused by the buildup of intracellular lipid droplets in liver cells, which can induce inflammation, cell death, and even more advanced stages such as nonalcoholic steatohepatitis (NASH) (with or without fibrosis), cirrhosis, and liver cancer [3, 4]. Currently, pharmacological options for NAFLD are limited. Treatment cornerstones are a healthy lifestyle and weight loss. There is still an unmet therapeutic need [5].
NAFLD is bidirectionally associated with components of the metabolic syndrome [6], a cluster of alterations that includes centripetal obesity, decreased HDL cholesterol concentrations, increased triglyceride concentrations, arterial hypertension, and hyperglycemia [7,8,9]. This syndrome has become one of the epidemics of the twenty-first century. Causative factors include insulin resistance, leptin, lipocalins, microbiota alterations, and epigenetics [10, 56]. In this regard, leptin should have an anti-steatosis impact on hepatocytes [15]. However, no therapeutics for NAFLD are directed at this target. As a result, the finding that leptin and NAFLD are correlated may be useful for assessing disease risk, preventing NAFLD, combining existing therapy regimens for potentiation, and identifying prospective targets for novel drug development. Our study, based on the literature, did find a significant association between increased leptin levels and reduced incidence of NAFLD, and our findings not only coincide with previous literature but also validate the hypothesis in the literature through database analysis of real-world case sources.
Leptin signals through binding to its receptors, mainly Lep Rb, which is a long stretch of extracellular structure, a transmembrane region and an elongated intracellular extension. As Lep Rb does not possess intrinsic kinase activity, the conformational change of Lep Rb upon leptin binding to Lep Rb induces the activation of Janus kinase (JAK2) phosphorylation, which phosphorylates three tyrosine residues (Y985, Y1077 and Y1138) in the intracellular extension of Lep Rb. These phosphorylated tyrosine residues then recruit proteins containing the SH2 phosphorylation recognition domain for downstream signaling. Currently, the most studied leptin signaling is the JAK/signal transducer and activator of transcription (STAT) pathway. Leptin and NAFLD exert their effects mainly through the JAK2/STAT3 pathway [57, 58]. An important role of leptin is to direct the storage of triglycerides in adipocytes and prevent their deposition in non-adipose tissues such as the liver, thus preventing hepatocyte lipotoxicity and apoptosis. Leptin also inhibits the production of hepatic glucose and the formation of new hepatic fat, acting as an insulin-like agent to prevent the development of NAFLD. Studies have shown that chronic central leptin infusion can reduce hepatic lipid synthesis gene expression and triglyceride levels by stimulating hepatic sympathetic activity and that this effect of leptin is associated with the PI3K signaling pathway, blocking which can specifically induce hepatic steatosis without causing obesity. In addition, leptin promotes fatty acid oxidation in the liver and increases fatty acid consumption in the liver [59]. In addition to its direct effects on the liver, leptin also affects hepatic glucose metabolism indirectly through its central regulation. Leptin infusion into the ventricles of type 1 diabetes mice inhibited the expression of glucagon, consistent with the phenotype of peripheral hyperleptinemia [60]. Specific expression of Lep Rb in the arcuate nucleus of the rat hypothalamus by adenoviral transfection improves peripheral insulin sensitivity and reduces hepatic gluconeogenesis in leptin receptor-deficient Koletsky rats [61]. The regulation of hepatic glucose by leptin may be related to the effect of its phosphatidylinositol 3-kinase (PI3K), which increases insulin signaling and decreases the expression of glucose synthesis genes such as glucose-6-phosphatase (G-6-P) and phosphoenolpyruvate carboxykinase (PEPCK) [61]. In addition, the effects of leptin can also be mediated by central neural regulation, e.g., selective severance of the hepatic vagus nerve can prevent hypothalamic leptin from regulating hepatic insulin sensitivity.
The mechanism of leptin in NAFLD has been supported by a large body of experimental data, and clinical studies on leptin and NAFLD have focused on the association of leptin or leptin receptor levels with NAFLD. The findings on circulating leptin levels in NAFLD patients are not very consistent, with some studies reporting high leptin expression in NAFLD patients [62, 63] and others finding no difference in leptin levels in NAFLD patients compared to non-NAFLD populations [64, 65]. Clinical studies of leptin and Lep R gene expression and SNPs in the NAFLD population have also been reported sporadically. Two small clinical studies showed no expression of the Lep R gene in liver tissue, while in peripheral leukocytes and abdominal adipose tissue Lep gene expression did not differ significantly between NAFLD patients and healthy populations [66, 67]. In another study, immunohistochemical staining of liver tissue for leptin showed that leptin expression was higher in patients with NAFLD than in the healthy population, consistent with altered circulating leptin levels [63]. Some of the Lep R gene SNP studies have also shown a positive association with the development of NAFLD, even if this association is not dependent on the presence of obesity. Given the complexity of clinical studies and the multilevel nature of clinical data, it is difficult to obtain direct evidence that leptin resistance causes NAFLD from the available clinical research data, which need to be interpreted with caution.
Our study has several advantages. First, the TSMR analysis method is based on the principle of Mendelian randomization-free segregation and combination, which excludes the influence of acquired factors (social environment and natural environment) on the study results at the genetic level. In order to successfully compensate for the vulnerability to confounding factors and reverse causality interference in traditional observational studies for inferring the etiology of complex disorders, the genes must arise prior to the disease with a precise causal time sequence [68, 69]. Second, this study uses publicly available GWAS summary statistics with a large sample size to obtain more precise estimates and greater statistical power, saving research costs and improving the utilization of biological information while limiting the study population mainly to individuals of European ancestry, reducing some of the bias that may arise due to population stratification. Finally, the value of this study lies in establishing an association between leptin levels and the incidence of NAFLD using a database of real-world sources. Based on our findings, it is reasonable to believe that leptin levels may be used for the assessment of NAFLD, including the screening of people who are traditionally at high risk of develo** NAFLD (e.g., those with comorbid diabetes and those who are overweight), and, more importantly, for the assessment of the risk of develo** lean NAFLD in people with normal BMI. In addition, it can be used as an indicator to evaluate the improvement potential of NAFLD. Since there are no drugs that target leptin, we believe that, based on the current state of research, leptin can reduce the incidence of NAFLD without duplicating the mechanism of action of other existing drugs for NAFLD and can be used as a complement to existing treatment regimens. Additionally, there is evidence that leptin regulation may have favorable effects on a variety of other factors, including weight loss, reducing blood sugar levels, and controlling intestinal functions [70]. Therefore, modulation of leptin levels may be used in multiple aspects of metabolic disorders and may have a wider range of potential applications. However, there are some limitations to this study. First and foremost, the majority of these GWAS data are from European populations. It needs to be determined if the findings we described would hold in other people. Second, this study lacks a multidimensional stratification of the heterogeneity of patients with NAFLD. In the future, a multicenter prospective cohort study is needed to fully consider the heterogeneity of NAFLD, integrate demographic characteristics, lifestyle, genetics and other factors to accurately identify high-risk groups for NAFLD, and develop targeted and individualized body mass control strategies, with a view to achieving accurate prevention and control of NAFLD.
Conclusion
In conclusion, our study reveals a causal relationship between leptin and NAFLD. It thus provides further insight into the factors that may be associated with a reduced risk of NAFLD development. Additionally, from a systems biology standpoint, it aids researchers in better understanding the connections between diverse diseases.
Data availability
All the relevant data are provided within the paper and are publicly available.
Abbreviations
- NAFLD:
-
Nonalcoholic fatty liver disease
- BMI:
-
Body Mass Index
- NASH:
-
Non-alcoholic steatohepatitis
- ALT:
-
Alanine transaminase
- MR:
-
Mendelian randomization
- TSMR:
-
Two-Sample Mendelian Randomization
- SNP:
-
Single Nucleotide Polymorphism
- GWAS:
-
Genome-Wide Association Studies
- IEU:
-
Integrated Epidemiology Unit
- IVs:
-
Instrumental variables
- IVW:
-
Inverse variance weighted
- WM:
-
Weighted Median
- OR:
-
Odds Ratio
- CI:
-
Confidence interval
- LD:
-
Linkage disequilibrium
- MAFLD:
-
Metabolic-dysfunction-associated fatty liver disease
References
Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, Wai-Sun Wong V, Yilmaz Y, George J, Fan J, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(6):2672–82.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84.
Yin X, Guo X, Liu Z, Wang J. Advances in the diagnosis and treatment of non-alcoholic fatty liver disease. Int J Mol Sci. 2023;24(3):2844.
Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. 2009;58(11):1538–44.
Paternostro R, Trauner M. Current treatment of non-alcoholic fatty liver disease. J Intern Med. 2022;292(2):190–204.
Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J Hepatol. 2018;68(2):335–52.
Neuschwander-Tetri BA. Non-alcoholic fatty liver disease. BMC Med. 2017;15(1):45.
Ren Z, Simons P, Wesselius A, Stehouwer CDA, Brouwers M. Relationship between NAFLD and coronary artery disease: a Mendelian randomization study. Hepatology. 2023;77(1):230–8.
Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20.
Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol. 2020;17(1):40–52.
Romeo S, Kozlitina J, **ng C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5.
Jiménez-Cortegana C, García-Galey A, Tami M, Del Pino P, Carmona I, López S, Alba G, Sánchez-Margalet V. Role of leptin in non-alcoholic fatty liver disease. Biomedicines. 2021;9(7):762.
Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: a narrative review. Metab Clin Exp. 2015;64(1):60–78.
Rotundo L, Persaud A, Feurdean M, Ahlawat S, Kim HS. The Association of leptin with severity of non-alcoholic fatty liver disease: a population-based study. Clin Mol Hepatol. 2018;24(4):392–401.
Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia. 2016;59(1):30–43.
Boutari C, Perakakis N, Mantzoros CS. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol Metab. 2018;33(1):33–43.
Adolph TE, Grander C, Grabherr F, Tilg H. Adipokines and non-alcoholic fatty liver disease: multiple interactions. Int J Mol Sci. 2017;18(8):1649.
Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, Sato N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 2001;34(2):288–97.
Zhang Q, Wang J, Huang F, Yao Y, Xu L. Leptin induces NAFLD progression through infiltrated CD8+ T lymphocytes mediating pyroptotic-like cell death of hepatocytes and macrophages. Digest Liver Dis. 2021;53(5):598–605.
Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33(1):30–42.
Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10(4):486–96.
Guo JZ, **ao Q, Gao S, Li XQ, Wu QJ, Gong TT. Review of mendelian randomization studies on ovarian cancer. Front Oncol. 2021;11: 681396.
Davey Smith G, Holmes MV, Davies NM, Ebrahim S. Mendel’s laws, Mendelian randomization and causal inference in observational data: substantive and nomenclatural issues. Eur J Epidemiol. 2020;35(2):99–111.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–6.
Richmond RC, Davey Smith G. Mendelian randomization: concepts and scope. Cold Spring Harb Perspect Med. 2022;12(1): a040501.
Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–52.
Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–26.
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–52.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7: e34408.
Yaghootkar H, Zhang Y, Spracklen CN, Karaderi T, Huang LO, Bradfield J, Schurmann C, Fine RS, Preuss MH, Kutalik Z, et al. Genetic studies of leptin concentrations implicate leptin in the regulation of early adiposity. Diabetes. 2020;69(12):2806–18.
Carter AR, Sanderson E, Hammerton G, Richmond RC, Davey Smith G, Heron J, Taylor AE, Davies NM, Howe LD. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465–78.
Ghodsian N, Abner E, Emdin CA, Gobeil É, Taba N, Haas ME, Perrot N, Manikpurage HD, Gagnon É, Bourgault J, et al. Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell Rep Med. 2021;2(11): 100437.
van Kippersluis H, Rietveld CA. Pleiotropy-robust Mendelian randomization. Int J Epidemiol. 2018;47(4):1279–88.
Chen L, Yang H, Li H, He C, Yang L, Lv G. Insights into modifiable risk factors of cholelithiasis: a Mendelian randomization study. Hepatology. 2022;75(4):785–96.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-98.
Yuan S, Chen J, Li X, Fan R, Arsenault B, Gill D, Giovannucci EL, Zheng JS, Larsson SC. Lifestyle and metabolic factors for nonalcoholic fatty liver disease: Mendelian randomization study. Eur J Epidemiol. 2022;37(7):723–33.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Allen NE, Sudlow C, Peakman T, Collins R. UK biobank data: come and get it. Sci Transl Med. 2014;6(224): 224ed224.
Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–89.
Huang S, Huang F, Mei C, Tian F, Fan Y, Bao J. Systemic lupus erythematosus and the risk of cardiovascular diseases: a two-sample Mendelian randomization study. Front Cardiovasc Med. 2022;9: 896499.
Li Q, Yan S, Li Y, Kang H, Zhu H, Lv C. Mendelian randomization study of heart failure and stroke subtypes. Front Cardiovasc Med. 2022;9: 844733.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, Hartwig FP, Holmes MV, Minelli C, Relton CL, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186.
Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64.
Eslam M, Sanyal AJ, George J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(7):1999-2014.e1991.
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, Zelber-Sagi S, Wai-Sun Wong V, Dufour JF, Schattenberg JM, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–9.
Ismaiel A, Jaaouani A, Leucuta DC, Popa SL, Dumitrascu DL. The visceral adiposity index in non-alcoholic fatty liver disease and liver fibrosis-systematic review and meta-analysis. Biomedicines. 2021;9(12):1890.
Nassir F. NAFLD: mechanisms, treatments, and biomarkers. Biomolecules. 2022;12(6):824.
Wong VW, Adams LA, de Lédinghen V, Wong GL, Sookoian S. Noninvasive biomarkers in NAFLD and NASH - current progress and future promise. Nat Rev Gastroenterol Hepatol. 2018;15(8):461–78.
Younossi ZM. Non-alcoholic fatty liver disease—a global public health perspective. J Hepatol. 2019;70(3):531–44.
Wang AY, Dhaliwal J, Mouzaki M. Lean non-alcoholic fatty liver disease. Clin Nutr. 2019;38(3):975–81.
Mantovani A, Dalbeni A. Treatments for NAFLD: state of art. Int J Mol Sci. 2021;22(5):2350.
Perakakis N, Farr OM, Mantzoros CS. Leptin in leanness and obesity: JACC state-of-the-art review. J Am Coll Cardiol. 2021;77(6):745–60.
Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci. 2011;16(7):2771–93.
Myers MG Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21(11):643–51.
Lee Y, Yu X, Gonzales F, Mangelsdorf DJ, Wang MY, Richardson C, Witters LA, Unger RH. PPAR alpha is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc Natl Acad Sci USA. 2002;99(18):11848–53.
Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA. 2010;107(40):17391–6.
German J, Kim F, Schwartz GJ, Havel PJ, Rhodes CJ, Schwartz MW, Morton GJ. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology. 2009;150(10):4502–11.
Lemoine M, Ratziu V, Kim M, Maachi M, Wendum D, Paye F, Bastard JP, Poupon R, Housset C, Capeau J, et al. Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int. 2009;29(9):1431–8.
Xu D, Huang XD, Yuan JP, Wu J, Fan Y, Luo HS, Yang YH. Impaired activation of phosphatidylinositol 3-kinase by leptin is a novel mechanism of hepatic leptin resistance in NAFLD. Hepatogastroenterology. 2011;58(110–111):1703–7.
Pagano C, Soardo G, Esposito W, Fallo F, Basan L, Donnini D, Federspil G, Sechi LA, Vettor R. Plasma adiponectin is decreased in nonalcoholic fatty liver disease. Eur J Endocrinol. 2005;152(1):113–8.
Kashyap SR, Diab DL, Baker AR, Yerian L, Bajaj H, Gray-McGuire C, Schauer PR, Gupta M, Feldstein AE, Hazen SL, et al. Triglyceride levels and not adipokine concentrations are closely related to severity of nonalcoholic fatty liver disease in an obesity surgery cohort. Obesity. 2009;17(9):1696–701.
Chalasani N, Crabb DW, Cummings OW, Kwo PY, Asghar A, Pandya PK, Considine RV. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am J Gastroenterol. 2003;98(12):2771–6.
Cayón A, Crespo J, Mayorga M, Guerra A, Pons-Romero F. Increased expression of Ob-Rb and its relationship with the overexpression of TGF-beta1 and the stage of fibrosis in patients with nonalcoholic steatohepatitis. Liver Int. 2006;26(9):1065–71.
Davey Smith G, Ebrahim S. What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ. 2005;330(7499):1076–9.
Tillmann T, Vaucher J, Okbay A, Pikhart H, Peasey A, Kubinova R, Pajak A, Tamosiunas A, Malyutina S, Hartwig FP, et al. Education and coronary heart disease: Mendelian randomisation study. BMJ. 2017;358: j3542.
Huang KP, Goodson ML, Vang W, Li H, Page AJ, Raybould HE. Leptin signaling in vagal afferent neurons supports the absorption and storage of nutrients from high-fat diet. Int J Obes. 2021;45(2):348–57.
Acknowledgements
We express our gratitude to https://gwas.mrcieu.ac.uk/ and https://www.ebi.ac.uk/gwas/ for providing publicly available summary-level GWAS data for leptin and NAFLD. In addition, the authors would like to thank all the reviewers who participated in the review and MJEditor ( www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82174341) and the New Teacher Start-up Fund project of the Bei**g University of Chinese Medicine (2022-JYB-XJSJJ-050).
Author information
Authors and Affiliations
Contributions
Study conception and design: ZG and JZ. Main data analysis and manuscript draft: HD and YG. Data analysis: QJ. Manuscript proofread: RL and ZY. Manuscript review: YY and XL. Study supervision and data analysis: ZG. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, Z., Du, H., Guo, Y. et al. Association between leptin and NAFLD: a two-sample Mendelian randomization study. Eur J Med Res 28, 215 (2023). https://doi.org/10.1186/s40001-023-01147-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01147-x