
Abstract
To improve the temperature characteristics and catalytic efficiency of a glycoside hydrolase family (GHF) 11 xylanase from Aspergillus oryzae (AoXyn11A), its variants were predicted based on in silico design. Firstly, Gly21 with the maximum B-factor value, which was confirmed by molecular dynamics (MD) simulation on the three-dimensional structure of AoXyn11A, was subjected to site-saturation mutagenesis. Thus, one variant with the highest thermostability, AoXyn11AG21I, was selected from the mutagenesis library, E. coli/Aoxyn11A G21X (X: any one of 20 amino acids). Secondly, based on the primary structure multiple alignment of AoXyn11A with seven thermophilic GHF11 xylanases, AoXyn11AY13F or AoXyn11AG21I–Y13F, was designed by replacing Tyr13 in AoXyn11A or AoXyn11AG21I with Phe. Finally, three variant-encoding genes, Aoxyn11A G21I, Aoxyn11A Y13F and Aoxyn11A G21I–Y13F, were constructed by two-stage whole-plasmid PCR method, and expressed in Pichia pastoris GS115, respectively. The temperature optimum (T opt) of recombinant (re) AoXyn11AG21I–Y13F was 60 °C, being 5 °C higher than that of reAoXyn11AG21I or reAoXyn11AY13F, and 10 °C higher than that of reAoXyn11A. The thermal inactivation half-life (t 1/2) of reAoXyn11AG21I–Y13F at 50 °C was 240 min, being 40-, 3.4- and 2.5-fold longer than those of reAoXyn11A, reAoXyn11AG21I and reAoXyn11AY13F. The melting temperature (T m) values of reAoXyn11A, reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F were 52.3, 56.5, 58.6 and 61.3 °C, respectively. These findings indicated that the iterative mutagenesis of both Gly21Ile and Tyr13Phe improved the temperature characteristics of AoXyn11A in a synergistic mode. Besides those, the catalytic efficiency (k cat/K m) of reAoXyn11AG21I–Y13F was 473.1 mL mg−1 s−1, which was 1.65-fold higher than that of reAoXyn11A.
Similar content being viewed by others

Introduction
Xylanase (endo-β-1,4-d-xylanase, EC 3.2.1.8) exclusively catalyzes the hydrolysis of internal β-1,4-d-xylosidic linkages in the xylan backbone, producing xylooligosaccharides via a double displacement mechanism in which one conserved Glu operates as a general acid catalyst and the other Glu does as a nucleophile (Trevizano et al. 2012). Based on the multiple alignment of primary structures and the analysis of hydrophobic clusters, most of xylanases have been classified into glycoside hydrolase families (GHFs) 10 and 11 (Luo et al. 2010). The overall three-dimensional (3-D) structure of GHF11 xylanases resembles a partially closed right hand, consisting mainly of one α-helix and two β-sheets which are packed against each other (Xue et al. 2012). In striking contrast with GHF10 xylanases, the GHF11 counterparts possessed the catalytic activities merely towards d-xylose-containing substrates, low molecular weights (MWs <30 kDa), and alkaline isoelectric points (pIs >7.0) although some eukaryotic xylanases were acidic pIs (Balaa et al. 2009).
To date, xylanases have been applied to a range of industrial bioprocesses, such as feed, food, textile, biofuel and papermaking (Irfan et al. 2016). However, the majority of wild-type xylanases displayed low thermostability, preventing them from being applied in bioprocesses where the high temperature was required and encountered, exemplified by enzyme-added feeds, baking and pulp bleaching (Kumar et al. 2016). A handful of thermophilic xylanases have been produced by thermophiles, but their specific activities and other enzymatic properties were very poor, making them unable to be applied effectively (Zhang et al. 2014). To meet the increasing demands for thermophilic xylanases, some modifications in the primary and/or 3-D structures of mesophilic counterparts with superior enzymatic properties were conducted by peptide segment substitution, site-directed mutagenesis, DNA shuffling and error-prone PCR (Song et al. 2014; Stephens et al. 2014; Watanabe et al. 2016; Zheng et al. 2014). In recent years, various mutagenesis techniques have been developed for the modifications of proteins/enzymes. For example, the site-saturation mutagenesis of Glu434 on the surface of RGI lyase was carried out to improve its thermostability. After experimental verification, the best variant of RGI lyase, Glu434Leu, was selected from the mutagenesis library. Its half-life at 60 °C (t 601/2 ) extended to 31 min, which was 1.6-fold longer than that of the wild-type lyase (Silva et al. 2013). For the site-directed or site-saturation mutagenesis, the selection of the amino acid positions or the substituted residues was a pivotal step, which could be predicted based on in silico design or computer-aided design (Yin et al. 2015).
In our previous study, an AoXyn11A-encoding gene, Aoxyn11A (GenBank accession No. JQ326257), was expressed in P. pastoris GS115. The reAoXyn11A displayed the high specific activity, broad pH stability and strong tolerance to metal ions, but low thermostability (Li et al. 2013). In this work, to improve the temperature characteristics and catalytic efficiency of AoXyn11A, its variants were were predicted based on in silico design. Then, the variant genes, Aoxyn11A G21I, Aoxyn11A Y13F and Aoxyn11A G21I–Y13F, were constructed by site-saturation and site-directed mutagenesis, and expressed in P. pastoris GS115, respectively. Finally, the enzymatic properties of recombinant variants were analyzed, and compared to those of reAoXyn11A. To our knowledge, this is the first report on the saturation mutagenesis and high-throughput screening (Gly21Ile) and the iterative mutagenesis (Gly21Ile and Tyr13Phe) of AoXyn11A based on in silico design to improve its enzymatic properties.
Materials and methods
Strains, vectors, and culture media
E. coli BL21(DE3) and pET-28a(+) (Novagen, Madison, WI, USA) were used for the construction and screening of a site-saturation mutagenesis library, while E. coli DH5α and pPIC9K (Invitrogen, San Diego, CA, USA) for the construction of recombinant expression vectors. The vectors pET-28a-Aoxyn11A and pPIC9K-Aoxyn11A, and transformants E. coli/Aoxyn11A and P. pastoris/Aoxyn11A were constructed in our lab (Li et al. 2013). The genes, Aoxyn11A and its variants, were expressed in P. pastoris GS115. E. coli BL21 and DH5α were grown in the LB medium (10 g L−1 tryptone, 5 g L−1 yeast extract and 10 g L−1 NaCl, pH 7.2). P. pastoris and its transformants were cultured and induced in the YPD, MD, geneticin G418-containing YPD, BMGY and BMMY media, which were prepared as described in the manual of Multi-Copy Pichia Expression Kit (Invitrogen, USA).
Multiple templates-based homology modeling
It was demonstrated that the validity of multiple templates-based modeling mainly relies on the primary structure identities of a target protein with template ones, and on the multiple alignment accuracy among crystal structures of templates (Madhusudhan et al. 2009). Compared to the single template-based modeling method, the multiple templates-based one greatly increased the facticity and accuracy of the modeled 3-D structure of a target protein (Sokkar et al. 2011). Therefore, using AoXyn11A primary structure as the template, homology sequences were searched among GHF11 xylanases at NCBI website (http://www.ncbi.nlm.nih.gov/) towards Protein Data Bank (PDB). The three known crystal structure xylanases separately from Penicillium funiculosum (PDB code: 1TE1) (Payan et al. 2004), Talaromyces cellulolyticus (3WP3) (Kataoka et al. 2014) and E. coli (2VUL) (Dumon et al. 2008), possessing the highest primary structure identities with AoXyn11A, were selected as homology modeling templates. The 3-D structures of AoXyn11A and its variants were modeled based on the three templates using the SALIGN program (http://salilab.org/DBAli/) and MODELLER 9.11 program (http://salilab.org/modeller/), and analyzed using the PyMol software (http://pymol.org/).
Prediction of the three variants of AoXyn11A
The 3-D structure of a mesophilic protein/enzyme was more flexible than that of the corresponding thermophilic analog (Jaenicke and Böhm 1998). Therefore, to quantify the protein/enzyme flexibility, the notion of B-factor values was introduced to reflect the smearing of atomic electron densities as a result of thermal motion and positional disorder. B-factor values, namely atomic displacement parameters, of amino acids, were generated by molecular dynamics (MD) simulation on the crystal or modeled 3-D structure of protein using the GROMACS 4.5 package (http://www.gromacs.org/) and analyzed using the B-FITTER program (Reetz and Carballeira 2007). In this work, the modeled 3-D structure of AoXyn11A was subjected to MD simulation at 300 K for 15 ns, followed by calculating the B-factor values of residues. Gly21 in AoXyn11A with the maximum B-factor value was confirmed, and then randomly substituted by site-saturation mutagenesis.
AoXyn11AY13F or AoXyn11AG21I–Y13F was designed by replacing Tyr13 in AoXyn11A or AoXyn11AG21I with Phe, based on the multiple alignment of AoXyn11A with seven representative thermophilic GHF11 xylanases using the ClustalW2 program (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Root mean square deviation (RMSD) value, which was defined as the Cα-atomic displacement range of a protein from its original configuration to changed one at a high temperature and a certain time, was negatively correlative to its thermostability (Badieyan et al. 2012). Thereby, to predict the thermostabilities of AoXyn11AY13F and AoXyn11AG21I–Y13F, the modeled 3-D structures of AoXyn11A, AoXyn11AY13F and AoXyn11AG21I–Y13F were subjected to MD simulations, respectively, at 500 K for 7 ns using the GROMACS 4.5 package, followed by calculating their RMSD values using a g_rms software in the GROMACS 4.5.
Construction and screening of the site-saturation mutagenesis library
The site-saturation mutagenesis of a Gly21-encoding codon in Aoxyn11A into any amino acid-encoding codon was performed using the two-stage whole-plasmid PCR technique (Sanchis et al. 2008). The PCR primers used for gene mutagenesis were listed in Additional file 1: Table S1. The recombinant vectors, pET-28a-Aoxyn11A G21X, were amplified from pET-28a-Aoxyn11A. In brief, the first-stage PCR amplification was carried out using a pair of PCR primers G21X-F (forward) and X11-R (reverse) under the following conditions: an initial denaturation at 98 °C for 3 min, followed by 30 cycles of at 98 °C for 10 s, 53 °C for 30 s and 72 °C for 45 s. Then, the second-stage PCR was performed using the first-stage PCR products as the primers: 30 cycles of at 98 °C for 10 s, 55 °C for 30 s and 72 °C for 5 min. The target PCR products (pET-28a-Aoxyn11A G21X) were digested by DpnI, and transformed into E. coli BL21, thereby constructing a site-saturation mutagenesis library (E. coli/Aoxyn11A G21X).
Single colonies (E. coli/Aoxyn11A G21X strains) were separately inoculated into 500 μL LB medium containing 50 μg mL−1 kanamycin in a 96-well plate, and cultured at 37 °C overnight as the seed culture. Then, the same medium was inoculated with 2% seed culture, and grown until OD600 reached 0.6. Expression of Aoxyn11A G21X was induced by 1 mM IPTG at 28 °C for 8 h. The cells were collected, suspended in 100 μL Na2HPO4–citric acid buffer (50 mM, pH 5.5), and disrupted by ultrasonic (650 W, 180 cycles of work for 2 s and rest for 8 s). The resulting supernatant was divided into two aliquots and added into two 96-well plates. To screen the mutagenesis library, one plate was treated at 60 °C for 20 min, while the other plate used as the control. Then, aliquots of 5 μL treated or untreated supernatant were correspondingly added into two new plates with 95 μL 5.0 mg mL−1 xylan in each well, incubated at 50 °C for 10 min and stopped by adding 50 μL DNS reagent. Thus, the positive strains were selected, by which the expressed recombinant xylanases retained over 80% of their original activities, and in which the mutant genes were DNA-sequenced. Furthermore, one strain expressing reAoXyn11AG21I with the highest thermostability, E. coli/Aoxyn11A G21I, was obtained by extending treating time to 30 min at 60 °C.
Site-directed mutagenesis of Aoxyn11A and Aoxyn11A G21I
The recombinant vector, pET-28a-Aoxyn11A G21I, was extracted from the obtained strain E. coli/Aoxyn11A G21I, and digested using EcoRI and NotI to release Aoxyn11A G21I. Then, Aoxyn11A G21I was inserted into pPIC9K vector digested using the same enzymes, followed by transforming it into E. coli DH5α. The recombinant vector, pPIC9K-Aoxyn11A G21I, was confirmed by DNA sequencing. Using pPIC9K-Aoxyn11A or -Aoxyn11A G21I as the template, the site-directed mutagenesis of Tyr13-encoding codon in Aoxyn11A or Aoxyn11A G21I into Phe-encoding codon was performed by two-stage whole-plasmid PCR using a pair of PCR primers Y13F-F (forward) and X11-R (reverse). The amplified target PCR products, that is, the recombinant vectors, pPIC9K-Aoxyn11A Y13F and -Aoxyn11A G21I–Y13F, were digested using DpnI, transformed into E. coli DH5α and confirmed by DNA sequencing.
Transformation of recombinant vectors and screening of P. pastoris transformants
Three recombinant vectors, pPIC9K-Aoxyn11A G21I, -Aoxyn11A Y13F and -Aoxyn11A G21I–Y13F, were linearized with SalI, and transformed into P. pastoris GS115, respectively, by electroporation using the Gene Pulser apparatus (Bio-Rad, Hercules, CA, USA). All P. pastoris transformants were primarily screened based on their abilities to grow on the MD plate, and then successively inoculated on the YPD plates containing G418 at concentrations of 1.0, 2.0 and 4.0 mg mL−1 for the screening of multiple copies of the integrated target gene. The P. pastoris transformant resisting high geneticin G418 concentration may have multiple copies of a gene, which can lead to the high expression of recombinant protein/enzyme (Invitrogen, USA). However, the expression level was not directly proportional to G418 concentration (Zhang et al. 2014). Thereby, a total of 60 P. pastoris transformants resistant to G418 of 4.0 mg mL−1, separately containing Aoxyn11A G21I, Aoxyn11A Y13F and Aoxyn11A G21I–Y13F, were picked out for expression tests.
Expression and purification of the recombinant xylanases
Expression of Aoxyn11A G21I, Aoxyn11A Y13F or Aoxyn11A G21I–Y13F in P. pastoris GS115 was performed according to the instruction of Multi-Copy Pichia Expression Kit (Invitrogen, USA) with slight modification. In brief, each single colony of 60 pastoris transformants was inoculated into 30 mL BMGY medium and cultured at 30 °C with 220 rpm until OD600 reached 2–4. Then, the cells were harvested by centrifugation, suspended in 30 mL BMMY medium and induced for the expression of recombinant xylanase by adding methanol to a final concentration of 1.0% at 24 h intervals at 30 °C for 72 h.
After expression, 10 mL supernatant containing the recombinant xylanase with a 6-His-tag at its C-terminus was loaded onto a nickel–nitrilotriacetic acid (Ni–NTA) column (Tiandz, Bei**g, China; 1 × 6 cm) equilibrated with buffer A (20 mM Tris–HCl, 500 mM NaCl and 20 mM imidazole, pH 7.9), followed by elution at a flow rate of 0.3 mL min−1 with buffer B as same as buffer A except for 250 mM imidazole. Aliquots of 1 mL eluent containing the target xylanase were pooled, dialyzed against 50 mM Na2HPO4–citric acid buffer (pH 5.5), and concentrated by ultrafiltration.
Enzyme activity and protein assays
Xylanase activity was assayed by measuring the amount of reducing sugars from birchwood xylan (Sigma, St. Louis, MO, USA) using the 3,5-dinitrosalicylic acid (DNS) method (Gao et al. 2013). One unit (U) was defined as the amount of xylanase liberating 1 μmol of reducing sugar equivalent per min under the assay conditions (at pH 5.5 and 50 °C for 10 min). The sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the method of Laemmli (1970). The separated proteins were visualized by staining with Coomassie Brilliant Blue R-250 (Sigma, USA) and apparent molecular weights of xylanases were estimated by comparison with the standard proteins using the Quantity One software. The protein concentration was measured with a BCA-200 Protein Assay Kit (Pierce, Rockford, IL).
pH characteristics of the recombinant xylanases
The pH optimum of purified recombinant xylanase was determined under the standard assay conditions, except 5.0 mg mL−1 birchwood xylan in 50 mM Na2HPO4–citric acid buffer at a pH range of 3.0–7.5. Aliquots of xylanase were incubated at 40 °C and varied pH values (Na2HPO4–citric acid buffer: pH 3.0–7.5; Tris–HCl buffer: pH 8.0–9.0) for 60 min. The pH stability in this work was defined as a pH range, over which the residual xylanase activity retained over 85% of its original one.
Temperature characteristics of the recombinant xylanases
The temperature optimum (T opt) of recombinant xylanase was measured, at pH optimum, at temperatures ranging from 40 to 70 °C. The inactivation half-life (t 1/2) of enzyme, a parameter for estimating its thermostability, was defined as a time, when the residual enzyme activity was 50%. In this work, to measure the t 501/2 of recombinant xylanase, aliquots of xylanase were incubated at 50 °C for different times.
The melting temperature (T m) was defined as a temperature, at which a protein configuration is half unfolded. The protein/enzyme with high T m meant it has a high thermostability (Jang et al. 2010). The T m of xylanase here was measured by protein thermal shift (PTS) method (Dong et al. 2016), using a PTS Kit (Applied Biosystems, Carlsbad, CA, USA) and a LightCycler 480II 96 Real-Time PCR system (Roche, Basel, Switzerland). Three replicates were conducted independently. The T m was a temperature which corresponds to the peak value of a derivative melting curve plotted using the ‘T m calling’ method.
Kinetic parameters of the recombinant xylanases
The reaction rate (U/mg) of birchwood xylan by xylanase was measured under the standard assay conditions, but the substrate concentrations from 1.0 to 10 mg mL−1. The reaction rate versus xylan concentration was plotted to verify whether the reaction mode conformed to the Michaelis–Menten equation. The kinetic parameters, K m and k cat, were determined by non-linear regression analysis using an Origin 9.0 software (http://www.originlab.com/). All data were expressed as the mean ± standard deviation (SD) from three independent replicates.
Results
Homology modeling of the xylanase 3-D structures
The primary structure identities of AoXyn11A with the three known crystal structure GHF11 xylanases from P. funiculosum (PDB code: 1TE1), T. cellulolyticus (3WP3) and E. coli (2VUL) were 72, 70 and 68%, respectively, suggesting that the crystal structures were suitable as templates. As a result, the 3-D structures of AoXyn11A (Additional file 1: Figure S1) and its variants were homologically modeled, respectively, based on the three templates by SALIGN and MODELLER 9.11 programs, and analyzed by PyMol software.
Identification of amino acid positions for mutagenesis
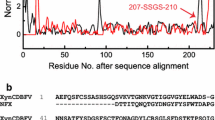
In this work, the B-factor values of amino acid residues in AoXyn11A, generated by MD simulation on its 3-D structure by GROMACS 4.5 package, were calculated using the B-FITTER program (Fig. 1). To improve the thermostability of AoXyn11A, Gly21, the amino acid numbered 21, with the maximum B-factor value of 67.75 Å was selected, and then subjected to site-saturation mutagenesis (Yin et al. 2015).

The B-factor values of amino acid residues in AoXyn11A. The modeled 3-D structure of AoXyn11A was subjected to MD simulation at 300 K for 15 ns. Gly21 in position 21 with the maximum B-factor value of 67.75 Å was marked
By consulting the literatures about GHF11 xylanases, the temperature characteristics of seven representative thermophilic GHF11 xylanases, sharing more than 50% identities with that of AoXyn11A, were summarized in Table 1. The multiple alignment of 8 or 11 xylanase primary structures displayed that Tyr13 in AoXyn11A (or in AoXyn11AG21I) corresponded to Phe in most thermophilic xylanases (Fig. 2; Additional file 1: Figure S2). As a result, the variant xylanase, AoXyn11AY13F or AoXyn11AG21I–Y13F was designed by substituting Tyr13 with Phe. Thereafter, RMSD values of AoXyn11A, AoXyn11AY13F (Fig. 3) and AoXyn11AG21I–Y13F were calculated. As shown in Fig. 3, the RMSD value of AoXyn11AY13F, after equilibration, was lower than that of AoXyn11A at any time. Thus, the designed AoXyn11AY13F or AoXyn11AG21I–Y13F was predicted to be more thermostable than AoXyn11A.

The multiple alignment of the N-terminal amino acid sequences of AoXyn11A and seven thermophilic GHF11 xylanases. The amino acids in positions 13 and 21, numbered by AoXyn11A, are highlighted in red solid line frame

The curves of RMSD values of AoXyn11A (black line) and AoXyn11AY13F (red line). The modeled 3-D structures of AoXyn11A and AoXyn11AY13F were subjected to MD simulations, respectively, at 500 K for 7 ns
Screening of the site-saturation mutagenesis library
For the primary screening of the saturation mutagenesis library of Gly21, all recombinant xylanases expressed by E. coli/Aoxyn11A G21X (X: any amino acid) were treated at 60 °C for 20 min. The results of activity assay and DNA sequencing displayed that seven recombinant variants (Gly21 mutated to Phe, Val, Leu, Arg, Tyr, His and Ile, respectively) retained more than 80% of their original activities (Additional file 1: Figure S3), but reAoXyn11A lost its activity completely. Then, four variants (reAoXyn11AG21F, reAoXyn11AG21V, reAoXyn11AG21L and reAoXyn11AG21I) were further screened by extending the treating time to 30 min, whose residual activities were 39.3, 30.5, 37.1 and 57.3%, respectively. Thus, the best strain E. coli/Aoxyn11A G21I was selected, expressing reAoXyn11AG21I with the highest thermostability.
Expression and purification of the recombinant xylanases
It was well known that the expression system of prokaryotes was simple in the gene manipulation and fast in the growth and induction expression rates. Compared to that of prokaryotes, the expression system of eukaryotes had some other advantages, such as extracellular expression, high purity and posttranslational modification, in the production of heterologous proteins/enzymes (Fernández et al. 2010). Based on the comparison of two kinds of expression systems, E. coli BL21(DE3) and pET-28a(+) (Novagen, USA) were applied for the construction and screening of a site-saturation mutagenesis library and for the DNA sequencing of a target gene in this work, while P. pastoris GS115 and pPIC9K (Invitrogen, USA) used for the expression and purification of recombinant reAoXyn11A and its variants. After P. pastoris transformant was induced by methanol for 72 h, the supernatant was used for xylanase activity and protein assays. Among 60 transformants tested, the three ones, labeled as P. pastoris/Aoxyn11A G21I4-1,/Aoxyn11A Y13F4-7 and/Aoxyn11A G21I–Y13F4-12 were selected, separately expressing the maximum reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F activities of 138.8, 89.4 and 79.1 U mL−1. Here, P. pastoris GS115 transformed with pPIC9K-Aoxyn11A, labeled as P. pastoris/Aoxyn11A, was used as the control.
Three recombinant variants, reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F, were purified to homogeneity by one-step way of Ni–NTA column chromatography. SDS-PAGE analysis of the three recombinant xylanases showed single protein bands with the same apparent molecular weight of about 22.1 kDa (Fig. 4, lanes 2–4). The specific activities of reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F under the standard assay conditions were 3677, 2280 and 1271 U mg−1, respectively, which were 2.3-, 1.4- and 0.8-fold higher than that (1593 U mg−1) of reAoXyn11A.

SDS-PAGE analysis of the purified reAoXyn11A and its variants. Lane M, protein marker; lane 1–4, reAoXyn11A, reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F purified from cultured supernatants, respectively
pH and temperature characteristics of the recombinant xylanases
The purified reAoXyn11A and its three variants exhibited the same pH optimum at pH 5.5 (Additional file 1: Figure S4a). Incubated at 40 °C and varied pH values (3.0–9.0) for 60 min, the four recombinant xylanases were stable over a broad pH range of 4.5–8.5, all retaining more than 85% of their original activities (Additional file 1: Figure S4b).
The T opt of reAoXyn11AG21I–Y13F was 60 °C, 5 °C higher than that of reAoXyn11AG21I or reAoXyn11AY13F and 10 °C higher than that of reAoXyn11A (Fig. 5a). The t 501/2 values of reAoXyn11AG21I, AoXyn11AY13F and reAoXyn11AG21I–Y13F were 71, 95 and 240 min, about 11.8-, 15.8- and 40-fold longer than that of reAoXyn11A (Fig. 5b). The emission intensity of the fluorescence dye combining with protein hydrophobic regions increased as the protein configuration was unfolded at high temperature (Niesen et al. 2007). Based on this mechanism, the T m values of reAoXyn11A, reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F were determined to be 52.3, 56.5, 58.6 and 61.3 °C, respectively (Fig. 6).

The temperature characteristics of four recombinant xylanases. a The T opt of xylanase was measured, at pH optimum, at temperatures ranging from 40 to 70 °C. b The t 501/2 of xylanase was assayed by incubated it at 50 °C for different times

The derivative melting curves of four recombinant xylanases. Emission intensities of fluorescence dye combining with protein hydrophobic regions were recorded from 30 to 80 °C at an elevated rate of 1 °C min−1
Analysis of the intramolecular interactions in xylanases
In this work, the differences of intramolecular interactions in AoXyn11A and AoXyn11AG21I–Y13F were analyzed using the PIC server (http://crick.mbu.iisc.ernet.in/PIC). The analytical findings indicated that two hydrophobic interactions (Pro3-Phe16 and Ile58-Val74) in AoXyn11A were demolished by substituting Gly21 with Ile, but nine additional hydrophobic interactions (Ile21-Ver41, Tyr34-Val172, Tyr71-Leu72, Ala73-Ile87, Val74-Val172, Tyr102-Ile115, Tyr113-Ile115, Ile115-Leu162 and Trp157-Tyr160) and one extra salt bridge (Glu84-Arg120) in AoXyn11AG21I–Y13F were generated (Fig. 7a). The mutation of Gly21Ile produced two large hydrophobic regions (Tyr34-Tyr71-Leu72-Ala73-Val74-Ile87-Val172 and Tyr102-Tyr113-Ile115-Trp157-Tyr160-Leu162) (Fig. 7b, c). The hydrophobic parameters of amino acids Tyr and Phe, which were positively correlative to their hydrophobicities, were -1.3 and 2.8, respectively (Thorton and Taylor 1989). Thus, it was confirmed that the hydrophobicity of Phe was more powerful than that of Tyr. Owing to the substitution of Tyr13 with Phe, three additional hydrophobic interactions (Phe13-Tyr8, Phe13-Tyr169 and Phe13-Ile171) presented in AoXyn11AG21I–Y13F were generated (Fig. 7d). These intramolecular interactions in the variants, AoXyn11AG21I, AoXyn11AY13F and AoXyn11AG21I–Y13F, played important roles in their improved temperature characteristics.

Analysis of the intramolecular interactions in AoXyn11AG21I–Y13F. a Nine extra hydrophobic interactions (Ile21-Val41, Tyr34-Val172, Tyr71-Leu72, Ala73-Ile87, Val74-Val172, Tyr102-Ile115, Tyr113-Ile115, Ile115-Leu162 and Trp157-Tyr160) and one additional salt bridge (Glu84-Arg120) were generated by substituting Gly21 with Ile. Three extra hydrophobic interactions (Phe13-Tyr8, Phe13-Tyr169 and Phe13-Ile171) were formed by mutation of Tyr13Phe. b, c Two large hydrophobic regions (Tyr34-Tyr71-Leu72-Ala73-Val74-Ile87-Val172 and Tyr102-Tyr113-Ile115-Trp157-Tyr160-Leu162) were illustrated in the locally magnified 3-D structures. d Three hydrophobic interactions (Phe13-Tyr8, Phe13-Tyr169 and Phe13-Ile171) were illustrated in the locally magnified 3-D structure
Kinetic parameters of the recombinant xylanases
The reaction mode of reAoXyn11A or its any recombinant variant conformed to the Michaelis–Menten equation. K m and k cat values of four recombinant xylanases towards birchwood xylan were determined and listed in Table 2. Compared with K m value (3.33 mg mL−1) of reAoXyn11A, those of reAoXyn11AG21I and reAoXyn11AG21I–Y13F were decreased by 30.3 and 37.5%, respectively, while that of reAoXyn11AY13F slightly increased by 4.2%. The k cat values of reAoXyn11A, reAoXyn11AG21I, reAoXyn11AY13F and reAoXyn11AG21I–Y13F were 954.1, 1246.8, 1137.5 and 984.1 s−1. Consequently, the catalytic efficiency, k cat/K m, of reAoXyn11AG21I–Y13F (473.1 mL mg−1 s−1) was higher than those (286.5 and 327.8 mL mg−1 s−1) of reAoXyn11A and reAoXyn11AY13F, but lower than that (537.4 mL mg−1 s−1) of reAoXyn11AG21I.
Discussion
Based on the permutation and combination of three continuous nucleotides or bases, the degenerate triad NNK (N: A/C/G/T; K: G/T) can constitute 32 codons (containing a terminator codon of TAG), which can encode all kinds of 20 amino acids. Therefore, NNK was commonly used for the site-saturation mutagenesis of a specific amino acid (Tian et al. 2013). The number of single colonies that should be screened for 95% coverage in the case of randomization at one amino acid position was 94, which could be predicted using the CASTER program (Yin et al. 2015). Therefore, more than 94 single colonies in this work were picked out from the site-saturation mutagenesis library of Gly21 in AoXyn11A (E. coli/Aoxyn11A G21X, X: any one of 20 residues) to select the best strain, which expressing the recombinant variant xylanase with the highest thermostability. Generally, based on their respective advantages, the prokaryote expression system was applied for the construction and screening of mutagenesis libraries, while the eukaryote one used for the production of recombinant proteins/enzymes.
For the site-directed and site-saturation mutagenesis, the identification of the amino acid residue sites or the substituted residues was a very inconvenient and time-consuming process. Fortunately, to date, a large amount of bioinformation about the relationship between protein’s structure and function has been calculated and analyzed by means of computer programs and softwares. Based on the computer-aided design, the key residues and their sites in the primary and 3-D structures, the substituted residues, and some properties of proteins/enzymes could be predicted more accurately and quickly (Lassila 2010). For example, some determinants for the improved thermostability of a mesophilic GHF11 xylanase, AuXyn11A, were predicted using the computational methods (Zhang et al. 2014). In this work, based on the 3-D structure homology modeling, MD simulation and B-factor value calculation, one specific amino acid and its position, namely Gly21, in AoXyn11A was confirmed for the site-saturation mutagenesis of Gly21X (X: any one of 20 amino acids) in AoXyn11A. Furthermore, based on the primary structure multiple alignment of AoXyn11A (or AoXyn11AG21I) with seven representative thermophilic GHF11 xylanases, and on the calculation and comparison of RMSD values of AoXyn11A and its variants, Tyr13 in AoXyn11A or AoXyn11AG21I was identified for the site-directed mutagenesis of Tyr13Phe.
Both amino acid residues, Tyr13 and Gly21, in AoXyn11A are located at its N-terminal region. Based on the modeling and analysis of the 3-D structure of AoXyn11A, it was identified that Tyr13 is located in the β-strand B2, while Gly21 lies in the loop between the β-strands B2 and A2 (Additional file 1: Figure S1). Unlike the C-terminus of GHF11 xylanase, which is buried in the center of two β-sheets A and B, its N-terminus lies in the fringe of two β-sheets and is exposed to the hydrophilic environment. Deduced from the above analytical results, the N-terminus is denatured more easily than the C-terminus at high temperature. The denaturation of GHF11 xylanase originated from its N-terminus, which would result in the disassembly of the whole 3-D structure of protein (Li et al. 2012). In our previous studies, N-terminus modifications of AoXyn11A were performed to improve its thermostability by different routes. The T opt and t 701/2 of reAEx11A, a hybrid xylanase constructed by substituting the N-terminus of AoXyn11A with the corresponding region of EvXyn11TS, was 30 °C higher and 197-fold longer than those of reAoXyn11A. But the V max of reAEx11A decreased to 804.4 μmol min−1 mg−1 (Gao et al. 2013). It was verified that the disulfide bridge in the N-terminus of AEx11A mainly contributed to its high thermostability (Tan et al. 2014). Besides, the T opt of reAoXyn11AC5/C32 was increased 10 °C by introducing a disulfide bridge between the β-strands A1 and A2 of AoXyn11A (Chen et al. 2013). In this work, both the temperature characteristics and catalytic efficiency of AoXyn11A were obviously improved by iterative mutagenesis of Gly21Ile and Tyr13Phe in the N-terminus.
Some minor or local modifications in the primary and 3-D structures related to the improved thermostability of GHF11 xylanases have been extensively reported, such as the increased number of charged residues, ion pairs and aromatic residues on the protein surface as well as the insertions to certain regions resulting in enhanced intramolecular interactions (You et al. 2010). These modifications or changes greatly increased the rigidity of a protein configuration, the property associated with its enhanced thermostability. Reportedly, several variants of Bacillus circulans xylanase with strengthened hydrophobic interactions in some local 3-D structures were much more thermostable than a wild-type xylanase (Joo et al. 2011). Similarly, the thermostability of xylanase XynR8 from Neocallimastigales rumen fungal was significantly improved by introducing several salt bridges into it (Xue et al. 2012). In this work, the structural analysis indicated that the additional hydrophobic interactions and a salt bridge in AoXyn11AG21I–Y13F make its N-terminus and whole 3-D structure more stable at high temperature, thereby obviously improving the thermostability of AoXyn11A.
In conclusion, based on the computer-aided design, the temperature characteristics and catalytic efficiency of a wild-type AoXyn11A were remarkably improved by site-saturation mutagenesis and iterative mutagenesis, making reAoXyn11AG21I–Y13F a promising candidate for industrial bioprocesses where the high temperature was required. This work also provided an effective strategy for the directed modification of other GHF11 xylanases, especially those from fungi, to perfect their enzymatic properties.
Abbreviations
- GHF11:
-
glycoside hydrolase family 11
- 3-D:
-
three-dimensional
- AoXyn11A:
-
GHF11 xylanase from Aspergillus oryzae
- Aoxyn11A :
-
AoXyn11A gene
- PDB:
-
Protein Data Bank
- MD:
-
molecular dynamics
- RMSD:
-
root mean square deviation
- IPTG:
-
isopropyl β-d-1-thiogalactopyranoside
- Ni–NTA:
-
nickel–nitrilotriacetic acid
- SDS-PAGE:
-
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- DNS:
-
3,5-dinitrosalicylic acid
- T opt :
-
temperature optimum
- t 1/2 :
-
thermal inactivation half-life
- T m :
-
melting temperature
- PTS:
-
protein thermal shift
- k cat/K m :
-
catalytic efficiency
References
Badieyan S, Bevan DR, Zhang C (2012) Study and design of stability in GH5 cellulases. Biotechnol Bioeng 109:31–44
Balaa BA, Brijs K, Gebruers K, Vandenhaute J, Wouters J, Housen I (2009) Xylanase XYL1p from Scytalidium acidophilum: site-directed mutagenesis and acidophilic adaptation. Bioresour Technol 100:6465–6471
Chen ZF, Zhang HM, Wang JQ, Tang CD, Wu J, Wu MC (2013) Engineering the thermostability of a xylanase from Aspergillus oryzae by an enhancement of the interactions between the N-terminus extension and the β-sheet A2 of the enzyme. Biotechnol Lett 35:2073–2079
Dong YH, Li JF, Hu D, Yin X, Wang CJ, Tang SH, Wu MC (2016) Replacing a piece of loop-structure in the substrate-binding groove of Aspergillus usamii β-mannanase, AuMan5A, to improve its enzymatic properties by rational design. Appl Microbiol Biotechnol 100:3989–3998
Dumon C, Varvak A, Wall MA, Flint JE, Lewis RJ, Lakey JH, Morland C, Luginbühl P, Healey S, Todaro T, DeSantis G, Sun M, Parra-Gessert L, Tan X, Weiner DP, Gilbert HJ (2008) Engineering hyperthermostability into a GH11 xylanase is mediated by subtle changes to protein structure. J Biol Chem 283:22557–22564
Fernández L, Jiao N, Soni P, Gumulya Y, de Oliveira LG, Reetz MT (2010) An efficient method for mutant library creation in Pichia pastoris useful in directed evolution. Biocatal Biotransfor 28:122–129
Gao SJ, Wang JQ, Wu MC, Zhang HM, Yin X, Li JF (2013) Engineering hyperthermostability into a mesophilic family 11 xylanase from Aspergillus oryzae by in silico design of N-terminus substitution. Biotechnol Bioeng 110:1028–1038
Hakulinen N, Turunen O, Jӓnis J, Leisola M, Rouvinen J (2003) Three-dimensional structures of thermophilic β-1,4-xylanases from Chaetomium thermophilum and Nonomuraea flexuosa: comparison of twelve xylanases in relation to their thermal stability. Eur J Biochem 270:1399–1412
Irfan M, Guler HI, Ozer A, Sapmaz MT, Belduz AO, Hasan F, Shah AA (2016) C-Terminal proline-rich sequence broadens the optimal temperature and pH ranges of recombinant xylanase from Geobacillus thermodenitrificans C5. Enzyme Microb Technol 91:34–41
Jaenicke R, Böhm G (1998) The stability of proteins in extreme environments. Curr Opin Struct Biol 8:738–748
Jang MK, Lee SW, Lee DG, Kim NY, Yu KH, Jang HJ, Kim S, Kim A, Lee SH (2010) Enhancement of the thermostability of a recombinant β-agarase, AgaB, from Zobellia galactanivorans by random mutagenesis. Biotechnol Lett 32:943–949
Joo JC, Pack SP, Kim YH, Yoo YJ (2011) Thermostabilization of Bacillus circulans xylanase: computational optimization of unstable residues based on thermal fluctuation analysis. J Biotechnol 151:56–65
Kataoka M, Akita F, Maeno Y, Inoue B, Inoue H, Ishikawa K (2014) Crystal structure of Talaromyces cellulolyticus (formerly known as Acremonium cellulolyticus) GH family 11 xylanase. Appl Biochem Biotechnol 174:1599–1612
Kumar V, Marín-Navarro J, Shukla P (2016) Thermostable microbial xylanases for pulp and paper industries: trends, applications and further perspectives. World J Microbiol Biotechnol 32:34–43
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lassila JK (2010) Conformational diversity and computational enzyme design. Curr Opin Chem Biol 14:676–682
Li H, Murtomäki L, Leisola M, Turunen O (2012) The effect of thermostabilising mutations on the pressure stability of Trichoderma reesei GH11 xylanase. Protein Eng Des Sel 25:821–826
Li JF, Gao SJ, Liu XT, Gong YY, Chen ZF, Wei XH, Zhang HM, Wu MC (2013) Modified pPIC9K vector-mediated expression of a family 11 xylanase gene, Aoxyn11A, from Aspergillus oryzae in Pichia pastoris. Ann Microbiol 63:1109–1120
Luo HY, Yang J, Li J, Shi PJ, Huang HQ, Bai YG, Fan YL, Yao B (2010) Molecular cloning and characterization of the novel acidic xylanase XYLD from Bispora sp. MEY-1 that is homologous to family 30 glycosyl hydrolases. Appl Microbiol Biotechnol 86:1829–1839
Madhusudhan MS, Webb BM, Marti-Renom MA, Eswar N, Sali A (2009) Alignment of multiple protein structures based on sequence and structure features. Protein Eng Des Sel 22:569–574
Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2:2212–2221
Payan F, Leone P, Porciero S, Furniss C, Tahir T, Williamson G, Durand A, Manzanares P, Gilbert HJ, Juge N, Roussel A (2004) The dual nature of the wheat xylanase protein inhibitor XIP-I: structural basis for the inhibition of family 10 and family 11 xylanases. J Biol Chem 279:36029–36037
Reetz MT, Carballeira JD (2007) Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc 2:891–903
Sanchis J, Fernández L, Carballeira JD, Drone J, Gumulya Y, Höbenreich H, Kahakeaw D, Kille S, Lohmer R, Peyralans JJ, Podtetenieff J, Prasad S, Soni P, Taglieber A, Wu S, Zilly FE, Reetz MT (2008) Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl Microbiol Biotechnol 81:387–397
Silva IR, Larsen DM, Jers C, Derkx P, Meyer AS, Mikkelsen JD (2013) Enhancing RGI lyase thermostability by targeted single point mutations. Appl Microbiol Biotechnol 97:9727–9735
Sokkar P, Mohandass S, Ramachandran M (2011) Multiple templates-based homology modeling enhances structure quality of AT1 receptor: validation by molecular dynamics and antagonist docking. J Mol Model 17:1565–1577
Song L, Dumon C, Siguier B, Andre I, Eneyskaya E, Kulminskaya A, Bozonnet S, O’Donohue MJ (2014) Impact of an N-terminal extension on the stability and activity of the GH11 xylanase from Thermobacillus xylanilyticus. J Biotechnol 174:64–72
Sriyapai T, Somyoonsap P, Matsui K, Kawai F, Chansiri K (2011) Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in Escherichia coli and Pichia pastoris. J Biosci Bioeng 111:528–536
Stephens DE, Khan FI, Singh P, Bisetty K, Singh S, Permaul K (2014) Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. J Biotechnol 187:139–146
Tan ZB, Tang CD, Wu MC, He Y, Hu D, Wang JQ (2014) Exploration of disulfide bridge and N-glycosylation contributing to high thermostability of a hybrid xylanase. Protein Pept Lett 21:657–662
Thorton J, Taylor WR (1989) Structure prediction. In: Findlay JBC, Geisow MJ (eds) The IRL Press series in protein sequencing. IRLPress, Oxford, pp 147–190
Tian J, Wang P, Huang L, Chu XY, Wu NF, Fan YL (2013) Improving the thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 using a computationally aided method. Appl Microbiol Biotechnol 97:2997–3006
Trevizano LM, Ventorim RZ, de Rezende ST, Junior FPS, Guimarães VM (2012) Thermostability improvement of Orpinomyces sp. xylanase by directed evolution. J Mol Catal B Enzym 81:12–18
Watanabe M, Fukada H, Ishikawa K (2016) Construction of thermophilic xylanase and its structural analysis. Biochemistry 55:4399–4409
Xue HP, Zhou JG, You C, Huang Q, Lu H (2012) Amino acid substitutions in the N-terminus, cord and α-helix domains improved the thermostability of a family 11 xylanase XynR8. J Ind Microbiol Biotechnol 39:1279–1288
Yin X, Li JF, Wang CJ, Hu D, Wu Q, Gu Y, Wu MC (2015) Improvement in the thermostability of a type A feruloyl esterase, AuFaeA, from Aspergillus usamii by iterative saturation mutagenesis. Appl Microbiol Biotechnol 99:10047–10056
You C, Huang Q, Xue H, Xu Y, Lu H (2010) Potential hydrophobic interaction between two cysteines in interior hydrophobic region improves thermostability of a family 11 xylanase from Neocallimastix patriciarum. Biotechnol Bioeng 105(5):861–870
Zhang F, Chen JJ, Ren WZ, Lin LB, Zhou Y, Zhi XY, Tang SK, Li WJ (2012) Cloning, expression, and characterization of an alkaline thermostable GH11 xylanase from Thermobifida halotolerans YIM 90462T. J Ind Microbiol Biotechnol 39:1109–1116
Zhang HM, Li JF, Wang JQ, Yang YJ, Wu MC (2014) Determinants for the improved thermostability of a mesophilic family 11 xylanase predicted by computational methods. Biotechnol Biofuels 7:3
Zhao LM, Geng J, Guo YQ, Liao XD, Liu XH, Wu RJ, Zheng ZJ, Zhang RJ (2015) Expression of the Thermobifida fusca xylanase Xyn11A in Pichia pastoris and its characterization. BMC Biotechnol 15:18
Zheng HC, Liu YH, Sun MZ, Han Y, Wang JL, Sun JS, Lu FP (2014) Improvement of alkali stability and thermostability of Paenibacillus campinasensis family-11 xylanase by directed evolution and site-directed mutagenesis. J Ind Microbiol Biotechnol 41:153–162
Authors’ contributions
XL and QW carried out the prediction of AoXyn11A’s variants, the site-saturation and site-directed mutagenesis of Aoxyn11A and the expression of its variant genes, and drafted the manuscript. DH, RW and YL performed the purification of recombinant xylanases and the analysis of enzymatic properties. JL and MW directed the overall study and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Authors are grateful to Prof. **anzhang Wu (School of Biotechnology, Jiangnan University, Wuxi, China) for providing technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article (and its Additional file 1).
Consent for publication
This article does not contain any individual person’s data.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This work was financially supported by the National Nature Science Foundation of China (No. 21676117) and the Postgraduate Innovation Training Project of Jiangsu, China (KYLX16_0804).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
13568_2017_399_MOESM1_ESM.pdf
Additional file 1: Table S1. PCR primers used for the site-saturation and site-directed mutagenesis. Figure S1. The modeled 3-D structure of AoXyn11A. Figure S2. The primary structure multiple alignment of AoXyn11A with three homology modeling templates and seven thermophilic GHF11 xylanases. Figure S3. The primary screening of the site-saturation mutagenesis library of Gly21 in AoXyn11A. Figure S4. The pH characteristics of our recombinant xylanases.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, XQ., Wu, Q., Hu, D. et al. Improving the temperature characteristics and catalytic efficiency of a mesophilic xylanase from Aspergillus oryzae, AoXyn11A, by iterative mutagenesis based on in silico design. AMB Expr 7, 97 (2017). https://doi.org/10.1186/s13568-017-0399-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-017-0399-9