
Abstract
Introduction
Metabolomic signatures of type 2 diabetes mellitus (T2DM) in Tibetan Chinese population, a group with high diabetes burden, remain largely unclear. Identifying the serum metabolite profile of Tibetan T2DM (T-T2DM) individuals may provide novel insights into early T2DM diagnosis and intervention.
Methods
Hence, we conducted untargeted metabolomics analysis of plasma samples from a retrospective cohort study with 100 healthy controls and 100 T-T2DM patients by using liquid chromatography–mass spectrometry.
Results
The T-T2DM group had significant metabolic alterations that are distinct from known diabetes risk indicators, such as body mass index, fasting plasma glucose, and glycosylated hemoglobin levels. The optimal metabolite panels for predicting T-T2DM were selected using a tenfold cross-validation random forest classification model. Compared with the clinical features, the metabolite prediction model provided a better predictive value. We also analyzed the correlation of metabolites with clinical indices and found 10 metabolites that were independently predictive of T-T2DM.
Conclusion
By using the metabolites identified in this study, we may provide stable and accurate biomarkers for early T-T2DM warning and diagnosis. Our study also provides a rich and open-access data resource for optimizing T-T2DM management.
Similar content being viewed by others


Introduction
Diabetes mellitus (DM) is a common chronic metabolic disease characterized by hyperglycemia resulting from insulin-omission [1]. Type 2 diabetes mellitus (T2DM), which accounts for more than 95% of all DM cases [2], is an important cause of diabetic complications and the high mortality in individuals with DM [3, 4]. Currently, approximately 462 million individuals suffer from T2DM worldwide, and China obtained roughly 24.4% (102.9 million) of all cases [5]. Recent surveys in China estimated that the overall prevalence of DM is 10.9%, and that of pre-DM is 35.7%. Among Tibetans, the age-standardized prevalence of DM and pre-DM was 6.2% and 19.7%, respectively, and continues to increase rapidly [6, 7].
The Tibetan Chinese population is one of the two largest human plateau-dwelling groups globally [8]. The risk factors of DM and the characteristics of glucose metabolism in Tibetans have already been extensively reported. For example, DM among Tibetans is associated with a higher annual family income, alcohol consumption, and higher fasting plasma glucose (FPG) level, independent of age, sex, and body mass index (BMI) [13], aromatic amino acids (phenylalanine and tyrosine) [13,14,15], other amino acids, acylcarnitines and certain lipids [16]. However, metabolomic signatures of incident DM among Tibetans remain largely unclear, and effective and reliable biomarkers for early T-T2DM diagnosis remain unknown. Considering the Tibetans’ particular glucose metabolism and genetic determinants of Tibetan high-altitude adaptation, evidence for the association of Tibetan T2DM (T-T2DM) with other amino acids or other types of metabolites is still very limited. Furthermore, insight into whether ethnic differences in these metabolite concentrations potentially contribute to the higher risk of T2DM remains uncertain. Therefore, we need to determine the metabolite concentrations, identify the associations with T-T2DM, and seek to establish ideal biomarkers for the early and accurate diagnosis of T-T2DM.
In the present study, we aimed to perform a metabolome-wide analysis of T2DM among Tibetans adults. We sought to reveal the clinical characteristics and metabolite signatures associated with T-T2DM. We found phenylalanine metabolism, phenylalanine, tyrosine and tryptophan biosynthesis, arachidonic acid metabolism as key disturbed pathways in T-T2DM. By employing machine learning and correlation analysis, we identified ten unique biomarkers and evaluated their diagnostic values for T-T2DM.
Research design and methods
Study design and population
In the current study, we recruited 100 patients from Hospital of Chengdu Office of People’s Government of Tibetan Autonomous Region (Hospital. C.T.) Sichuan, China, between 31 and 2020 and 30 October 2021. Our study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board for Clinical Research of Hospital. C.T. A comprehensive battery of surveys and a clinical assessment with fasting blood draw were conducted by trained, certified, and bilingual staff at in-person clinic visits from October 2020 to October 2021. The study was approved by the institutional review boards at all participating institutions, and all participants gave written informed consent. Inclusion criteria include (1) 20–75 years old; (2) diagnostic criteria of 2-DM (an fasting plasma glucose (Glu0) ≧ 7.0mmol/l and/or a 2-h blood glucose (Glu120) level ≧ 11.1mmol/l and/or an haemoglobin A1c (HbA1c) ≧ 6.5%, all patients); (3) Blood pressure below 140/90 mmHg; and (4) signed informed consent. Exclusion criteria include (1)T1DM or other specific types of diabetes mellitus; (2) acute complications of DM; (3) complication of serious primary diseases in cardiovascular, cerebrovascular, liver, kidney, and the hematopoietic system as well as a tumor; (4) suffering from mental illness and unable to cooperate; (5) pregnant or lactating women, or those preparing for pregnancy, women in their menstrual period; (6) recent use of psychoactive drugs or hormones; and (7) those who have participated in other clinical trials within the past 1 month.
Healthy controls, who participated in yearly health screenings during the study period and had no clinical evidence of glaucoma or a family history of glaucoma, were also consecutively enrolled from Hospital. C.T. Exclusion criteria of healthy controls: any hematopoietic system disorders, any hepatobiliary diseases, any coagulation abnormalities, taking medications that can affect blood cell components or serum biochemistry profiles, any systemic diseases (such as hypertension, diabetes, infections, systemic autoimmune diseases, and cancers), or any other neurodegenerative disorders. According to the inclusion and exclusion criteria, a total of 100 healthy controls were included (Fig. 1A). Demographics and clinical parameters of patients with T2DM (n = 100) and healthy controls (n = 100) were shown in Table 1.

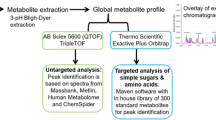
Study design. (A) The flowchart of patients enrolled in this study. T1DM, type 1 diabetes mellitus; DM, diabetes mellitus; T-T2DM, Tibetan type 2 diabetes mellitus; T-HC, Tibetan healthy controls. (B) Overview of workflow in this study. LC-MS, liquid chromatography-mass spectrometry
Figure 1B provides a detailed workflow of the metabolomics study. First, we collected 200 serum samples from the two groups and used metabolomics to identify the metabolite biomarkers of T-T2DM. We then used the metabolites identified by differential expression analyses to test machine learning models. This analysis highlighted potential biomarkers for clinical diagnosis and pathways involved in T-T2DM onset and progression.
Measurements of blood glucose and covariates
We collected information regarding gender, age, lifestyle factors, medical history, sociodemographic characteristics, and family history using a standardized questionnaire [17]. Systolic blood pressure (SBP) and diastolic blood pressure (DBP) and waist circumference were performed following standardized protocols [18]. Body mass index (BMI) was calculated as weight in kilograms divided by height in meters squared. Centralized laboratory tests were performed to determine plasma fasting glucose and hemoglobin A1c (HbA1c), and serum uric acid, creatinine, liver enzymes and lipids including triglycerides, and total, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) cholesterol. For T-T2DM participants, 2-h plasma glucose levels were also measured following a standard 75-g 2-h oral glucose tolerance test (OGTT).
Sample collection
Participants were asked to fast for at least 8 h before the examination, consume only water and necessary medications, and to refrain from smoking or physical activity before undergoing the fasting examination procedures. Venous blood samples were collected, processed, and frozen (at -80 °C) on-site toward the beginning of the visit.
Serum metabolomics profiling by liquid chromatography mass spectrometry (LC-MS) (LC-MS)
The prepared samples were analyzed using an ultraperformance HPLC (UHPLC) system (1290, Agilent Technologies) with a UPLC HSS T3 column (2.1 mm × 100 mm, 1.8 mm, Waters) coupled to Q Exactive Focus (Thermo Fisher Scientific, MA, USA), via a previously described method with some modifications [19]. Additional details are provided in the Supplementary Material Methods section.
Machine learning prediction on metabolomics data
For each pairwise comparison, based on the features selected by the differential metabolite analysis, random forest classification was applied by R and Bioconductor packages ‘random forest’ and ‘ggplot2’. The random forest classification (RFC) for identifying T-T2DM was trained on 140 randomly selected subjects (67 healthy subjects, 73 with T-T2DM), and then tested on the remaining subjects (33 healthy subjects, 27 with T-T2DM). The analysis was conducted with 5 repetitions of 10-fold cross-validation, using cross-validation error curves to select features as described elsewhere [20]. To improve the sensitivity of the integrated biomarker profiling (IBP) prediction model for T-T2DM, analysis of variance, Mean Decrease in Accuracy, and Gini impurity were used to rank potential biomarkers by importance [21]. The Receiver operating characteristic (ROC) curves and area under the curve (AUC) were calculated by R package ‘pROC’. The AUC, accuracy, sensitivity, specificity, and precision were used to evaluate the model performance. Detailed methods are in the Supplementary Material Methods section.
Data analysis
The clinical characteristics of patients were compared using the Fisher’s exact test for categorical variables and the Wilcoxon rank-sum test for continuous variables. These metabolites were annotated using the KEGG database (https://www.genome.jp/kegg/pathway.html), Human Metabolome Database (HMDB) (https://hmdb.ca/ metabolites) and LIPID MAPS Structure Database (http://www.lipidmaps.org/). Principal components analysis (PCA) and Partial least squares discriminant analysis (PLS-DA) were performed at metaX [22]. We applied univariate analysis (t-test) to calculate the statistical significance (P-value). The metabolites with VIP > 1 and P-value < 0.05 and fold change ≥ 1.2 or FC ≤ 0.833 were considered to be differential metabolites. Clustering heat maps were plotted by Pheatmap package in R language. Volcano plots were performed by ggplot2 in R. The correlation between metabolites and clinical characteristics were analyzed by corrplot package in R (method = pearson), P-value < 0.05 was considered as statistically significant. The metabolic pathways were considered as enrichment by impact values, when P-value of metabolic pathway < 0.05, metabolic pathway was considered as statistically significant enrichment.
The variables were selected based on variable importance in the projection (VIP > 1.0) from the peak height. In addition to the multivariate statistical method, Student’s t-test was also applied to measure the significance of each lipid. The resultant p values for each metabolite in all cross-comparisons were corrected by the Bonferroni correction. The resultant P values from ANOVA were further adjusted by the false discovery rate (FDR) based on the Hochberg-Benjamini method. Significantly altered variables were defined and further identified by VIP > 1.0, P < 0.05, and FDR < 0.05.
Statistics
All the statistical analyses were performed by statistical software R (R version R-3.4.3) with corresponding packages available. P-value ≤ 0.05 (or FDR = 5% for multiple hypothesis testing) was used to define significance.
Result
Cohort characteristics, sample collection
Table 1 enumerates the characteristics of the Tibetan participants. We included 100 patients with T-T2DM (40 females and 60 males) and 100 healthy controls (T-HC; 50 females and 50 males). The median (interquartile range, IQR) age of all participants was 47 (32–54) years. The T-T2DM cohort aged 32–65 years old, with a BMI of 18.20–38.97 kg/m2. Factors such as sex, BMI, low-density lipoprotein cholesterol level, and total cholesterol level were not significantly different between the T-T2DM and T-HC groups. The T-T2DM group was more likely to be hypercholesterolemic and had significantly higher triglyceride levels and lower serum uric acid and creatinine levels than the T-HC group. The mean values for FPG and 2-hour oral glucose tolerance test in the T-T2DM group were 7.90 ± 2.08 and 9.19 ± 1.16 mmol/L, respectively. Clearly, the T-T2DM group had higher levels of FPG and glycosylated hemoglobin (HbA1c) than the T-HC group.
Individual metabolites and risk of diabetes
To identify the serum metabolome features of the patients in the T-T2DM and T-HC groups, we generated untargeted metabolome profiles from fasting serum samples by means of LC-MS. Figure 2 A shows the median expression of 30 differentially expressed metabolites in the two groups. Both the principal components analysis score plot (Fig. 2B) and orthogonal partial least squares discriminant analysis (OPLS-DA model: R2Y(cum) = 0.88, Q2Y(cum) = 0.85, Fig. 2C), which were validated by permutation tests (200 permutations), revealed significant metabolite differences between the T-T2DM and T-HC groups. Overall, 1369 metabolites have been detected in serum. Among them, 412 (30.09%) significantly correlated with incident T-T2DM. Furthermore, 236 of these 412 metabolites largely included lipids and lipid-like molecules (11.41%), organic acids and derivatives (8.74%), and organoheterocyclic compounds (7.77%), and 14.8% were within 6 other metabolites (Fig. 2D). Of these 412 significant metabolites, 32 were positively associated with DM risk, while the 380 remaining metabolites showed an inverse association (Fig. 2E). To identify differentially expressed metabolites by pairwise comparisons, we conducted a nonparametric Wilcox rank-sum test on each metabolite (Supplementary Tables 1 and Fig. 2F). The metabolites were mainly components of amino acid metabolism and lipid metabolism. The top 5 metabolites showing upregulated expression were glutamine-asparagine-lysine (QNK), phenylalanine-proline-lysine (FPK), cyclo (glycyltryptophylprolylglycylvalylglycyl-β-hydroxytyrosyl), thymopentin and 6beta-Naltrexol-d3. Conversely, N-methyloctan-1-amine, quercetin, 2-(4,4-diphenyl-1-piperidinobuta-1,3-dienyl) phenyl acetate, phenylalanine-proline-histidine (FPH) and N,N’-di[4-(2,6-dimethylmorpholino)phenyl]thiourea were the top 5 metabolites demonstrating downregulated expression.

Detection of differentially expressed metabolites by pairwise comparison of T-T2DM. (A) Hierarchical clustering of differentially expressed metabolites. The median expression levels of metabolites (n = 30) for two groups are presented in the heatmap. (B) Score plots from the PCA model derived from the UPLC-MS profile of serum in two groups. (C) Score plots from the OPLS-DA model from metabolic profiles of two groups. (D) Pie-chart for the classification of significant differentially expressed metabolites (n = 412) according to meta-intensity. (E) Volcano plot of significantly differentially expressed metabolites with marking the top 5 up and down expression differential metabolites (Red represents up-regulated, blue represents down-regulated metabolites). (F) Representative box plots for top up-regulated and down-regulated metabolites
Correlation network of differential metabolites in serum
To explore the impact of metabolite alterations on T-T2DM, we conducted pathway and network analyses. As shown in Fig. 3A, we generated bubble plots to illustrate top significant pathways enriched by these biomarkers for each pairwise comparison. In the pathway analyses, phenylalanine metabolism; phenylalanine, tyrosine, and tryptophan biosynthesis; arachidonic acid metabolism, and riboflavin metabolism were superior. Consistently, our network analyses showed many key metabolites linked to T-T2DM development, including L-phenylalanine, phenylpyruvate, 2-hydroxyphenylacetate, arachidonate, and acetoacetate, and they were significantly altered in the T-T2DM and T-HC groups (Supplementary Tables 2 and Fig. 3B).

Correlation network of differential metabolites in serum. (A) KEGG analysis of significant functional pathways involved according to the differentially expressed metabolites. (B) Network analysis based on the top 10 KEGG pathways and their differential metabolites. The edges indicate the correlations between metabolites and metabolites, the size of node indicates the improtance of pathway (Red nodes represents metabolites, blue nodes represents pathways)
Machine learning for the pairwise predictions of T-T2DM from serum metabolite expression
For verifying the values of the identified metabolites in predicting T-T2DM status, we established the random forest classification (RFC) model to investigate whether metabolic profiling could predict DM development in Tibetans, independent of the primary diagnostic criteria of DM (Glu0, Glu120, and HbA1c). Initially, the predictive performance of metabolites for T-T2DM prediction was examined using randomly selected participants classified into the training (n = 140) and validation datasets (n = 60). The area under the receiver operating characteristics (ROC) curve (AUC) was 99.0% (95% CI: 97.4–100%, Fig. 4A). Then, we assessed the individual contribution of each feature to the classification accuracy via the random forest variable importance analysis for each class in the two models by the Mean Decrease in Accuracy and Gini impurity, which indicates the importance of the feature for the classification performance. Figure 4B-C illustrates the relative importance of the 20 most predictive metabolites. The top 5 predictive metabolites were 4-acetyl-4-(ethoxycarbonyl)heptanedioic acid, threonine-histidine-cysteine (THC), (±)12(13)-DiHOME, N-methyloctan-1-amine, and 2-methylbutyl beta-D-glucopyranoside by Gini importance. Supplementary Fig. 1 shows the paired differences in metabolite concentration between two groups. Moreover, we generated ROC curves from a fivefold cross-validation for all metabolites and successfully constructed a model containing the five most predictive metabolites (Fig. 4D). The model exhibited an AUC of 99.9% for T-T2DM prediction (Fig. 4E). Its prediction performance was estimated, and the T-T2DM group in the training and validation sets could be more broadly separated than the T-HC (Fig. 4F). group. We also calculated the AUCs of the traditional diagnostic metrics (BMI, fasting glucose and HbA1c, AUC = 0.5871, 95%CI = 0.5078–0.6664, AUC = 0.9138, 95%CI = 0.8669–0.9607, AUC = 0.9747, 95%CI = 0.9534–0.9960, respectively, Fig. 4G) and compared the prediction performance of T-T2DM risk factors such as age, triglycerides, serum uric acid, and creatinine (Fig. 4H). Together, these results indicated that the metabolite prediction model provided a better predictive value than the clinical features.

Establishment of integrated biomarker profiling. (A) AUC of the integrated 412 differential metabolites based on random forest classification (RFC) model. (B) the Mean Decrease in Accuracy (MDA) of 20 potential biomarkers. (C) Gini impurity of 20 potential biomarkers. (D) Distribution of 5 trials of 10-fold cross-validation error in random forest classifiers. The model was trained with 412 differential metabolites in the training set (T-T2DM group, n = 73; diabetes group, n = 67). The black solid curve showed the trials. The red line indicated the number of picked features in the optimal set. (E) AUC of the 5 selected potential biomarkers from the RFC model. (F) The prediction performance of the model consisted of 5 potential biomarkers in the train and test sets. (G) ROC curves for traditional markers BMI, fasting glucose and HbA1c. (H) ROC curves for risk factors of T-T2DM (age, triglycerides, serum uric acid and creatinine)
Potential biomarker panel discovery for predicting T-T2DM
Given that the diagnostic value of serum metabolites for T-T2DM remains unknown, we systemically analyzed correlations between top 50 predictive metabolites of Gini importance and the clinical parameters of each patient (Supplementary Table 3). Impressively, most of the metabolites negatively correlated with age, triglycerides, FPG, and HbA1c, Meanwhile, four metabolites, namely, 3-(2-methylpropyl)-octahydropyrrolo[1,2-a]pyrazine-1,4-dione, FPK, vincristine, cyclo(glycyltryptophylprolylglycylvalylglycyl-β-hydroxytyrosyl) and thymopentin were significantly associated with increased T-T2DM risk (Fig. 5A). Furthermore, the predictive ability of T-T2DM was analyzed by ROC analysis. As shown in Fig. 5B, the top 5 predictive metabolites of Gini importance, especially THC, (±)12(13)-DiHOME and N-methyloctan-1-amine, had a stronger predictive power than FPG. These five metabolites were also identified as potentially predictive markers for T-T2DM (Fig. 5C). We then assessed pairwise Pearson correlations between levels of these metabolites. The top 5 decreased metabolites and four increased metabolites showed strong mean correlations (r = 0.59, Fig. 5D). Collectively, changes in serum metabolites mentioned above may be effective biomarkers for determining T-T2DM onset and progression.

Potential biomarkers panel discovery for predicting T-T2DM. (A) A heat map shows the correlation between the top 50 metabolites of Gini impurity and clinical features (Red asterisks represent a positive correlation and blue asterisks represent a negative correlation). (B) ROC curves for each of the 5 selected potential biomarkers from the RFC model. (C) ROC curves for the five increased metabolites of the top 50 metabolites of Gini impurity. (D) The heat map illustrates Pearson correlations between potential biomarkers panel (Red circles, positive correlation; blue circles, negative correlation, the number inside each circle is Pearson correlation coefficient)
Discussion
Few studies have specifically reported the clinical characteristics of T2DM in Tibetan Chinese population. Hence, this retrospective cohort study described the detailed clinical features of Tibetans with T2DM in comparison with those of the healthy population. We found that the age, higher triglyceride, FPG, and HbA1c levels, and lower serum uric acid and creatinine levels were significant risk factors of T-T2DM. These risk factors were equally likely to develop long-term cardiovascular and kidney diseases [23]. The association of factors such as age [24], hypertriglyceridemia [25], and FPG [26],and HbA1c levels [27] with the risk of T2DM has been established. However, the association of the serum uric acid and creatinine levels with T2DM presence remains undetermined. Serum uric acid has been reported to be negatively associated with FPG, HbA1c, and high-density lipoprotein cholesterol [28, 29]. Other studies demonstrated lower serum uric acid level in the context of hyperglycemia, similar to our results [30, 31, 29], In addition, reduced serum creatinine levels were significantly associated with increased T2DM risk [32, 33]. This association could be explained by hyperfiltration of glomeruli and ectopic accumulation of adipose tissue combined with low muscle mass [34,35,36].
Physiological changes have been identified in Tibetans living at high altitudes, and many studies have unveiled the genetic bases of these physiological changes and a distinct metabolic signature for this population [37]. Tibetans may be vulnerable to glucose intolerance, with polycythemia as an indication of hypoxia adaptation [38]. Tibetans reportedly have higher oxidative stress than the Han counterparts, and a higher oxidative stress is associated with glucose intolerance and arteriosclerosis [39, 40]. The hypoxia-inducible factor pathway and metabolic features such as low cardiac phosphocreatine-to-ATP ratios, increased cardiac glucose uptake, and lower muscle mitochondrial densities have been observed in the high-altitude–adapted Tibetan native population [41]. These unique features contribute to Tibetans’ distinct metabolic changes.
To our knowledge, this study is the first to report novel predictive metabolic markers and altered metabolic profiles of T2DM among Tibetans in China. Significantly high levels of aromatic amino acids and BCAAs (leucine, isoleucine, and valine), low carbon number lipids (myristic, palmitic, and stearic acid), and significantly reduced pyroglutamic acid, glycerophospohlipids, and sphingomyelins are associated with T2DM [42, 43]. In the current study, the amino acids phenylalanine, tyrosine, and tryptophan were downregulated in the T-T2DM group compared with those in the HC group. Moreover, BCAA derivatives such as 4-hydroxyisoleucine (VIP value = 1.012, Q-value < 0.001, fold-change (T-T2DM/T-HC) = 0.689) and N-acetylvaline (VIP value = 1.145, Q-value < 0.001, fold-change (T-T2DM/T-HC) = 0.794) were downregulated in the T-T2DM group. In particular, 4-hydroxyisoleucine is useful for DM treatment because of its capacity for increasing insulin secretion [44]. Conversely, the levels of tripeptide compounds including QNK and FPK were increased in the T-T2DM group, and their increase was associated with DM risk. Further, β-hydroxybutyric acid (BHA) is a ketone body that has been described as an early biomarker of DM or diabetic ketoacidosis (DKA) [45, 46]. In DKA, increased free fatty acid oxidation and acidosis will lead to reduced mitochondrial redox state (nicotinamide adenine dinucleotide plus hydrogen-to-NAD1 ratio), promoting BHA production. Consistent with these results, the BHA level in our study (VIP value = 2.186, Q-value < 0.001, fold-change (T-T2DM/T-HC) = 1.499) was significantly higher in the T-T2DM group than in the T-HC group. Monitoring serum BHA levels may help early diagnose T-T2DM and detect DKA.
In network analyses, the metabolomic signatures were associated with phenylalanine metabolism; phenylalanine, tyrosine, and tryptophan biosynthesis; arachidonic acid metabolism, and riboflavin metabolism. Phenylalanine stimulates insulin secretion and further regulates compensatory mechanisms in the early stages of insulin resistance. Once compensated insulin secretion is met, individuals could subsequently progress to overt T2DM. Our results of metabolite changes in T-T2DM are consistent with recent findings in which decrease in aromatic amino acid levels occur after T2DM progression [47, 48]. In addition, the T-T2DM group had significantly reduced levels of free arachidonate and prostaglandin H2 in arachidonic acid metabolism; this significant decrement may also be a cause of glucose and lipid metabolism disorders in this group [49].
Several prospective metabolomic studies have investigated T2DM risks and biomarkers in a Chinese population by machine learning [48, 50]. A validated integrated biomarker profiling (IBP) was constructed using amino acids, L-carnitine, and acetyl-L-carnitine for the prediction of impaired fasting glucose and T2DM disease risks [50]. The present study used RFC to select the top 5 biomarkers of Gini impurity that have a good prediction ability of T-T2DM disease risk for IBP construction. These five biomarkers were 4-acetyl-4-(ethoxycarbonyl)heptanedioic acid, THC, (±)12(13)-DiHOME, N-methyloctan-1-amine, and 2-methylbutyl beta-D-glucopyranoside. The predicted performance of the model was satisfactory and better than the traditional markers of T2DM. These predictive metabolites negatively correlated with age, triglycerides, FPG, and HbA1c. Of note, (±)12(13)-DiHOME, an adipokine from brown adipose tissue, is closely related to the homeostasis of blood glucose and the metabolism balance of fatty acids and other lipids [51]. In addition, (±)12(13)-DiHOME is a peroxisome proliferator-activated receptor-γ receptor agonist that lowers blood glucose by enhancing systemic insulin sensitivity [52, 53]. However, (±)12(13)-DiHOME deletion in T-T2DM individuals could potentially contribute to a tightly linked interplay of increased oxidative stress and reduced insulin secretion, resulting in hyperglycemia secondary to inability to compensate for reduced insulin sensitivity.
This study has some limitations that should be considered. First, this study included patients from one institution only, and the sample size is small. Second, to capture a large number of metabolites, we used an untargeted metabolomic approach that could not measure the absolute values of metabolites. Nevertheless, this limitation did not impede our ability to estimate the associations between metabolites and the risk of T-T2DM. Third, some unmeasured factors (e.g., changes in lifestyle factors, or other diseased states over time) might have influenced our findings. Hence, our prospective study results should be interpreted with caution. Last, we only applied one machine learning method. More machine learning methods and further deep mining or algorithms may be needed. And our machine learning model was validated in the same cohort of subjects. We need to increase the sample volume for validating the results in an external cohort.
In conclusion, this study systematically profiled wide-ranging serum metabolites that were found to be associated with DM risk in Tibetan adults. Through metabolomics and a machine learning method, we have established the IBPs of T-T2DM and discovered potential biomarkers for predicting T-T2DM. Our findings may provide valuable diagnostic tools for the clinical implementation and design of effective novel therapeutic targets to achieve earlier T-T2DM prevention, diagnosis, and treatment.
Data Availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Abbreviations
- T-T2DM:
-
Tibetan with type 2 diabetes mellitus
- DM:
-
Diabetes mellitus
- T2DM:
-
Type 2 diabetes mellitus
- FPG:
-
Fasting plasma glucose
- BMI:
-
body mass index
- LC-MS:
-
liquid chromatography mass spectrometry
- SBP:
-
Systolic blood pressure
- DBP:
-
Diastolic blood pressure
- HbA1c:
-
Hemoglobin A1c, LDL:Low-density lipoprotein
- HDL:
-
High-density lipoprotein
- OGTT:
-
Oral glucose tolerance test
- T-HC:
-
Tibetan healthy controls
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- IBP:
-
Integrated biomarker profiling
References
Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–8.
Edwards KS, Ashraf S, Lomax TM, et al. Uncoupling protein 3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res Cardiol. 2018;113:47.
[Anonymous]. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. The Lancet Diabetes & Endocrinology. 2014;2:634–47.
Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40.
Khan MAB, Hashim MJ, King JK, et al. Epidemiology of type 2 diabetes - global burden of Disease and Forecasted Trends. J Epidemiol Glob Health. 2020;10:107–11.
Wang L, Gao P, Zhang M, et al. Prevalence and ethnic pattern of diabetes and Prediabetes in China in 2013. JAMA. 2017;317:2515–23.
Wang L, Peng W, Zhao Z, et al. Prevalence and treatment of diabetes in China, 2013–2018. JAMA. 2021;326:2498–506.
Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in tibetan and andean populations using dense genome scan data. PLoS Genet. 2010;6:e1001116.
Shaopeng Xu QW, **anjia Ning J, Liu, Wang J. The prevalence of and risk factors for diabetes mellitus and impaired glucose tolerance among Tibetans in China: a crosssectional study. 2017; 8: 112467–76.
Zhao Y, Yao Z, D’Souza W, et al. An epidemiological survey of stroke in Lhasa. Tibet China Stroke. 2010;41:2739–43.
Clish CB. Metabolomics: an emerging but powerful tool for precision medicine. Cold Spring Harb Mol Case Stud. 2015;1:a000588.
Satheesh G, Ramachandran S, Jaleel A. Metabolomics-Based prospective studies and prediction of type 2 diabetes Mellitus Risks. Metab Syndr Relat Disord. 2020;18:1–9.
Wang TJ, Larson MG, Vasan RS, et al. Metabolite profiles and the risk of develo** diabetes. Nat Med. 2011;17:448–53.
Alena Stan cáková MC, Niyas K, Saleem P, Soininen AJ. Kangas,3 Henna Cederberg,1 Jussi Paananen,1 Jussi Pihlajamäki,5 Lori L. Bonnycastle,6 Mario A. Morken,6 Michael Boehnke,7 Päivi Pajukanta,8 Aldons J. Lusis,2 Francis S. Collins,6 Johanna Kuusisto,1 Mika Ala-Korpela,3,4,9 and Markku Laakso. Hyperglycemia and a common variant of GCKR are Associated with the levels of eight amino acids in 9,369 finnish men. Diabetes. 2012;61:1895–902.
Anna Floegel NS, Mühlenbruch ZYuK. Schulze,4 Jerzy Adamski,6 Heiner Boeing,1 and Tobias Pischon1,1. Identification of Serum Metabolites Associated With Risk of Type 2 Diabetes Using a Targeted Metabolomic Approach. Diabetes. 2013;62:639–48. Dagmar Drogan,1 Hans-Georg Joost,5 Andreas Fritsche,2 Hans-Ulrich Häring,2 Martin Hrab e de Angelis,6 Annette Peters,7 Michael Roden,8,9 Cornelia Prehn,6 Rui Wang-Sattler,3 Thomas Illig,3,10.
Wang TJ, Ngo D, Psychogios N, et al. 2-Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest. 2013;123:4309–17.
Ning G, Reaction Study G. Risk evaluation of cAncers in chinese diabeTic individuals: a lONgitudinal (REACTION) study. J Diabetes. 2012;4:172–3.
American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32(Suppl 1):62–7.
Wang J, Zhang T, Shen X et al. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS. Metabolomics 2016; 12.
Feng Q, Liang S, Jia H, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528.
Abeles J, Conway DJ. The Gini coefficient as a useful measure of malaria inequality among populations. Malar J. 2020;19:444.
Wen B, Mei Z, Zeng C, et al. metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinformatics. 2017;18:183.
Jhund PS, Solomon SD, Docherty KF, et al. Efficacy of Dapagliflozin on renal function and outcomes in patients with heart failure with reduced ejection fraction: results of DAPA-HF. Circulation. 2021;143:298–309.
Li S, Guo S, He F, et al. Prevalence of diabetes mellitus and impaired fasting glucose, associated with risk factors in rural Kazakh adults in **njiang, China. Int J Environ Res Public Health. 2015;12:554–65.
Guerrero-Romero F, Rodriguez-Moran M. Hypertriglyceridemia is associated with development of metabolic glucose disorders, irrespective of glucose and insulin levels: a 15-year follow-up study. Eur J Intern Med. 2014;25:265–9.
Han Y, Zhang S, Chen S et al. Incidence and risk factors of type 2 diabetes mellitus in individuals with different fasting plasma glucose levels. 2020; 11: 2042018820928844.
Juarez DT, Demaris KM, Goo R, et al. Significance of HbA1c and its measurement in the diagnosis of diabetes mellitus: US experience. Diabetes Metab Syndr Obes. 2014;7:487–94.
Li W, Wang Y, Ouyang S, et al. Association between serum uric acid level and carotid atherosclerosis and metabolic syndrome in patients with type 2 diabetes Mellitus. Front Endocrinol (Lausanne). 2022;13:890305.
Hairong N, Zengchang P, Shaojie W, et al. Serum uric acid, plasma glucose and diabetes. Diab Vasc Dis Res. 2010;7:40–6.
Choi HK, Ford ES. Haemoglobin A1c, fasting glucose, serum C-peptide and insulin resistance in relation to serum uric acid levels–the Third National Health and Nutrition Examination Survey. Rheumatology (Oxford). 2008;47:713–7.
Ioachimescu AG, Brennan DM, Hoar BM, et al. Serum uric acid, mortality and glucose control in patients with type 2 diabetes mellitus: a PreCIS database study. Diabet Med. 2007;24:1369–74.
Song DK, Hong YS, Sung YA, et al. Association of serum creatinine levels and risk of type 2 diabetes mellitus in Korea: a case control study. BMC Endocr Disord. 2022;22:4.
Takeuchi M, Imano H, Muraki I et al. Serum creatinine levels and risk of incident type 2 diabetes mellitus or dysglycemia in middle-aged japanese men: a retrospective cohort study. Vet Rec 2018; 6.
Solerte SB, Gazzaruso C, Bonacasa R, et al. Nutritional supplements with oral amino acid mixtures increases whole-body lean mass and insulin sensitivity in elderly subjects with sarcopenia. Am J Cardiol. 2008;101:69E–77E.
Tonneijck L, Muskiet MH, Smits MM, et al. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol. 2017;28:1023–39.
Umegaki H. Sarcopenia and diabetes: hyperglycemia is a risk factor for age-associated muscle mass and functional reduction. J Diabetes Investig. 2015;6:623–4.
Simonson TS, McClain DA, Jorde LB, et al. Genetic determinants of tibetan high-altitude adaptation. Hum Genet. 2012;131:527–33.
Okumiya K, Sakamoto R, Ishimoto Y, et al. Glucose intolerance associated with hypoxia in people living at high altitudes in the tibetan highland. BMJ Open. 2016;6:e009728.
Sakamoto R, Matsubayashi K, Kimura Y, et al. Comprehensive geriatric assessment of elderly highlanders in Qinghai, China, III: oxidative stress and aging in Tibetan and Han elderly highlanders. Geriatr Gerontol Int. 2009;9:352–8.
Sakamoto R, Okumiya K, Wang H, et al. Oxidized low density lipoprotein among the Elderly in Qinghai-Tibet Plateau. Wilderness Environ Med. 2015;26:343–9.
Murray AJ. Energy metabolism and the high-altitude environment. Exp Physiol. 2016;101:23–7.
Zhao J, Zhu Y, Hyun N, et al. Novel metabolic markers for the risk of Diabetes Development in American Indians. Diabetes Care. 2014;38:220–7.
Muilwijk M, Goorden SMI, Celis-Morales C et al. Contributions of amino acid, acylcarnitine and sphingolipid profiles to type 2 diabetes risk among south-asian surinamese and dutch adults. BMJ Open Diabetes Res Care 2020; 8.
Zafar MI, Gao F. 4-Hydroxyisoleucine: a potential New treatment for type 2 diabetes Mellitus. BioDrugs. 2016;30:255–62.
Stojanovic V, Ihle S. Role of beta-hydroxybutyric acid in diabetic ketoacidosis: a review.
Fujii S, Maeda T, Noge I, et al. Determination of acetone in saliva by reversed-phase liquid chromatography with fluorescence detection and the monitoring of diabetes mellitus patients with ketoacidosis. Clin Chim Acta. 2014;430:140–4.
Mahendran Y, Jonsson A, Have CT, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia. 2017;60:873–8.
Ren M, Lin DZ, Liu ZP, et al. Potential novel serum metabolic markers Associated with Progression of Prediabetes to overt diabetes in a Chinese Population. Front Endocrinol (Lausanne). 2021;12:745214.
Sonnweber T, Pizzini A, Nairz M et al. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int J Mol Sci 2018; 19.
Long J, Yang H, Yang Z, et al. Integrated biomarker profiling of the metabolome associated with impaired fasting glucose and type 2 diabetes mellitus in large-scale chinese patients. Clin Transl Med. 2021;11:e432.
Jaikanth C, Gurumurthy P, Indhumathi T, et al. Emergence of SFRP5 as a pleiotropic adipocytokine and its association with wnt signaling pathways. Minerva Endocrinol. 2017;42:280–9.
Derosa G, Sahebkar A, Maffioli P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J Cell Physiol. 2018;233:153–61.
Nieman DC, Shanely RA, Luo B, et al. Metabolomics approach to assessing plasma 13- and 9-hydroxy-octadecadienoic acid and linoleic acid metabolite responses to 75-km cycling. Am J Physiology-Regulatory Integr Comp Physiol. 2014;307:R68–R74.
Acknowledgements
This research was funded by the Science and Technology Project of Tibet Autonomous Region: grant numbers XZ202102YD0026D and XZ202201ZR0037G, the central government guides local projects (Fund No. XZ202102YD0032C), the Science and technology project of Sichuan Province (Fund No. 2021YJ0161), the Medical Research project of Sichuan Province (Fund No. Q20042), Hospital level key project of Hospital of Chengdu office of People’s Government of Tibetan Autonomous Region (Fund No. 2021-YJ-2). We also acknowledged NOVOGENE Company Limited (Bei**g, China).
Funding
This research was funded by the Science and Technology Project of Tibet Autonomous Region: grant numbers XZ202102YD0026D and XZ202201ZR0037G, the central government guides local projects (Fund No. XZ202102YD0032C), the Science and technology project of Sichuan Province (Fund No. 2021YJ0161), the Medical Research project of Sichuan Province (Fund No. Q20042), Hospital level key project of Hospital of Chengdu office of People’s Government of Tibetan Autonomous Region (Fund No. 2021-YJ-2).
Author information
Authors and Affiliations
Contributions
JM, FH, JC and YW contributed to the study conception and design. JM, FH, JS, CZ, LF, SW and HL performed material preparation, experiments, data collection and analysis. YG, XH, XL and WH analyzed data. The frst draft of the manuscript was written by JM and FH. JC and YW reviewed and revised manuscript. JS, CZ, LF and SW reviewed the paper and gave suggestions on the revision of the article. JC and YW provided fnancial supports. All authors read and approved the fnal manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the Institutional Review Board for Clinical Research and Animal Ethics Committee of Hospital of Chengdu Office of People’s Government of Tibetan Autonomous Region (Hospital. C.T.).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Meng, J., Huang, F., Shi, J. et al. Integrated biomarker profiling of the metabolome associated with type 2 diabetes mellitus among Tibetan in China. Diabetol Metab Syndr 15, 146 (2023). https://doi.org/10.1186/s13098-023-01124-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13098-023-01124-8