
Abstract
Background
Angiostrongyliasis is a highly dangerous infectious disease. Angiostrongylus cantonensis larvae migrate to the mouse brain and cause symptoms, such as brain swelling and bleeding. Noncoding RNAs (ncRNAs) are novel targets for the control of parasitic infections. However, the role of these molecules in A. cantonensis infection has not been fully clarified.
Methods
In total, 32 BALB/c mice were randomly divided into four groups, and the infection groups were inoculated with 40 A. cantonensis larvae by gavage. Hematoxylin and eosin (H&E) staining and RNA library construction were performed on brain tissues from infected mice. Differential expression of long noncoding RNAs (lncRNAs) and mRNAs in brain tissues was identified by high-throughput sequencing. The pathways and functions of the differentially expressed lncRNAs were determined by Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses. The functions of the differentially expressed lncRNAs were further characterized by lncRNA‒microRNA (miRNA) target interactions. The potential host lncRNAs involved in larval infection of the brain were validated by quantitative real-time polymerase chain reaction (qRT‒PCR).
Results
The pathological results showed that the degree of brain tissue damage increased with the duration of infection. The transcriptome results showed that 859 lncRNAs and 1895 mRNAs were differentially expressed compared with those in the control group, and several lncRNAs were highly expressed in the middle–late stages of mouse infection. GO and KEGG pathway analyses revealed that the differentially expressed target genes were enriched mainly in immune system processes and inflammatory response, among others, and several potential regulatory networks were constructed.
Conclusions
This study revealed the expression profiles of lncRNAs in the brains of mice after infection with A. cantonensis. The lncRNAs H19, F630028O10Rik, Lockd, AI662270, AU020206, and Mexis were shown to play important roles in the infection of mice with A. cantonensis infection.
Graphical Abstract

Similar content being viewed by others

Background
Parasitic diseases, especially helminth infections, which are among the most important neglected tropical diseases (NTDs), pose a serious threat to humans globally [1]. More than 2 billion people worldwide are infected with helminths, resulting in more than 12 million disability-adjusted life years (DALYS) [2, 3].
For example, the global prevalence of Ascariasis is reported to be 446 million infections, hookworms cause 173 million infections, schistosomiasis affects more than 200 million people in 74 countries, and the World Health Organization estimates that globally, more than 600 million people are infected by Strongyloides stercoralis [4,5,6]. Angiostrongylus cantonensis is a zoonotic parasitic nematode with a complex indirect life cycle [7, 8] and is one of the most common pathogens causing eosinophilic meningoencephalitis [9]. The third-stage larvae of A. cantonensis are its infective form [10], and its life cycle is completed mainly in rats and snails; gastropods, such as slugs and snails, are its intermediate hosts and mammals, such as rats, are its final hosts [11]. More than 20 vertebrate species, including humans, may serve as accidental hosts, while amphibians and fish may serve as transfer hosts [12,13,14]. A. cantonensis may develop into adult nematodes in rats only; mice and humans are nonpermissive hosts [15]. A. cantonensis cannot mature into worms in mice. Humans are mainly infected by raw or semiraw consumption of pathogen-carrying fruits and vegetables and undercooked intermediate hosts [16]. Early symptoms of A. cantonensis infection includes nausea, vomiting, breathing difficulties, headaches, and low-grade fever. Later, the infection progresses to severe chronic headache, paralysis, and even coma or death [17].
The disease is prevalent mainly in Southeast Asia, the Caribbean, the Pacific basin, and other regions [18]. In recent years, the global number of cases of infection with A. cantonensis has increased annually [9]. The disease burden of A. cantonensis is likely underestimated [11]. In China, the first case of human infection with A. cantonensis was reported in Taiwan in 1944 when A. cantonensis was found in the cerebrospinal fluid of a young man exhibiting meningeal symptoms and eosinophils in the cerebrospinal fluid [19]. From 1945 to 2008, a total of 769 cases were reported in China, accounting for approximately 27.22% of the global number of cases and posing a serious threat to people’s health [16].
Noncoding RNAs (ncRNAs) are RNAs that do not encode proteins and account for approximately 98% of the human genome [20, 21]. NcRNAs include ribosomal RNA (rRNA), transfer RNA (tRNA), long noncoding RNA (lncRNA), circular RNA (circRNA), microRNA (miRNA), etc.; lncRNAs are conserved RNAs with a length of > 200 nucleotides [22, 23]. With the flourishing development of high-throughput technologies, the role of lncRNAs in the growth and development of living organisms as well as in disease processes has gradually been revealed. LncRNAs are key genetic regulators of different biological processes and are involved in regulating epigenetic regulation, cell differentiation, the cell cycle, and immune response [24]. An increasing number of studies have shown that lncRNAs can act as competitive endogenous RNAs that bind to miRNAs and participate in various biological processes [25, 26].
A growing emphasis has been placed on zoonotic diseases with the idea of “One Health” [27], of which A. cantonensis is one of the most important emerging diseases. At present, the main treatment for angiostrongyliasis is the use of anthelmintic drugs, which can relieve symptoms and reduce disease duration. However, this treatment may also lead to the release of intracellular contents from dying worms to increase the inflammatory response. Moreover, lncRNAs show promise as novel biomarkers and therapeutic targets for various diseases [28]. Consequently, for better prevention and treatment of angiostrongyliasis and to interrupt transmission of the disease, this study used a BALB/c mouse model to mimic a human infection model to screen differentially expressed lncRNAs in the brain tissue of infected mice and validate their dynamic changes during the course of the infection. This study will provide therapeutic targets and new diagnostic protocols for the treatment of angiostrongyliasis. In addition, the functions of the differentially expressed lncRNAs were further identified by lncRNA‒miRNA target interactions.
Methods
Mouse model establishment and hematoxylin and eosin (H&E) staining
Achatina fulica were originally obtained from Kai**, Guangdong, China, and the lung tissues were dissected, isolated, and ground. The third-stage larvae of A. cantonensis were removed from the ground homogenate. Thereafter, 32 6–8 week-old BALB/c female rats (Shanghai Jihui Co., Ltd.) were randomly divided into a negative control group and an infected group at a ratio of 3:5. Each mouse in the experimental group was gavaged with 40 third-stage larvae, and the mice in the negative control group were gavaged with saline. After larval infection, brain tissues were collected at 1, 7, 14, and 21 days, fixed in 4% paraformaldehyde, embedded in paraffin, dewaxed in xylene, immersed in different concentrations of ethanol and stained with H&E. Pathological changes in the brains of infected mice at different infection times were observed through H&E staining. After H&E staining, the nuclei were stained blue, and the cytoplasm was stained pink [29].
Ribonucleic acid extraction library construction
Total RNA was extracted from mouse brain tissue samples at 14 days after infection using TRIzol reagent, RNA purity was assessed, RNA quantification was performed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA), and RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples that passed quality control were used for subsequent library construction. Ribosomal RNA was removed using the Ribo-off rRNA Depletion Kit (Vazyme, Nan**g, China). The transcriptome library was constructed using the VAHTS Universal V6 RNA-seq Library Prep Kit according to the instructions. The whole transcriptome was sequenced and analyzed by Shanghai Ouyi Biotechnology Co.
RNA sequencing and differentially expressed gene analysis
The libraries were sequenced using the Illumina NovaSeq 6000 sequencing platform, and 150 bp bipartite reads were generated. Approximately 125,994 M raw reads were obtained from each sample. Raw reads in fastq format were processed using fastp software, and clean reads were obtained by removing low-quality reads for subsequent data analysis [30]. Negative control group comparisons were performed using HISAT2 software [31]. The read counts for each gene were obtained by HTSeq-count [32], and gene expression (FPKM) calculations were performed to select differentially expressed genes [33].
Differentially expressed gene analysis was performed using DESeq2 software, where genes that met the thresholds of q value (adjusted P value) < 0.05 and fold change > 2 or fold change < 0.5 were defined as differentially expressed genes (DEGs) [34]. Hierarchical clustering analysis of DEGs was performed using R (v 3.2.0) to demonstrate the expression patterns of genes across samples and groups. Subsequently, Gene Ontology (GO) and Kyoto Gene and Genome Encyclopedia (KEGG) pathway enrichment analyses of DEGs based on hypergeometric distribution algorithms were used to screen for significantly enriched functional entries [35, 36].
Quantitative real-time PCR (qPCR) validation
In total, six DE lncRNAs were selected for qRT‒PCR analyses to validate the DEG‒Seq results and their dynamics during infection. Reverse transcription was performed using a reverse transcription system kit (TaKaRa, Japan). The primer sequences are shown in Additional file 1, with GAPDH serving as the internal reference primer. Quantitative real-time PCR (qRT‒PCR) analysis was performed using SYBR® Green Real-Time Fluorescent Quantitative PCR Premix (TaKaRa, Japan).
Protein‒protein interaction (PPI) network construction
The STRING database (http://string-db.org/) was used to predict interactions between proteins, and a combined PPI score greater than 0.4 for differentially expressed mRNAs was used as a critical value [29]. Network map** of the relationships of the top 50 DE mRNAs was performed on the basis of interaction score sorting.
LncRNA–mRNA interaction study
The correlation between the six samples in the infected group and the negative control group was calculated using the Pearson correlation test. The correlation analysis set a threshold of an absolute value of the correlation coefficient greater than or equal to 0.80 and a P value less than or equal to 0.05. Differentially expressed lncRNAs and genes from the same differential comparison group were identified using Circos plotting software [37].
A hypergeometric distribution test was utilized to identify the miRNAs with the greatest impact among the differentially expressed lncRNAs. For the total differential lncRNA enrichment results, the top 300 miRNA‒lncRNA interaction pairs with smaller P values were selected in order of P value, and the R network package was used to map the lncRNA‒miRNA targets [38].
Statistical analysis
Statistical analysis was performed using GraphPad Prism ver. 8.0.2 (GraphPad Software, Inc., San Diego, CA, USA). The expression level of each gene was represented as an FC according to the 2–△△Ct method. The Student’s t test was used to analyze the differences between the groups. All the data are expressed as the mean ± standard deviation. All experiments were performed on no fewer than three biological replicates. Significance was defined as a P value < 0.05 [Full size image

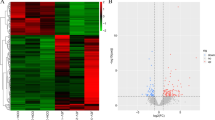
Differentially expressed mRNA profiles. Differential mRNA grou** clustering graph (A): the graph indicates relatively high expression mRNAs in red and relatively low expression mRNAs in blue. Differential expression volcano graph (B): the differences resulting from the comparison are reflected in the volcano graph, with nonsignificantly different mRNAs in gray and significantly different mRNAs in red and green; the horizontal axis is log2FoldChange, and the vertical axis is −log10 P Value
GO analysis and KEGG pathway analysis
The expression of many lncRNAs/mRNAs was significantly dysregulated in mice infected with A. cantonensis 14 days after infection. To reveal the function of aberrant lncRNAs/mRNAs, we performed GO and KEGG pathway enrichment analyses.
The top 10 GO terms were sorted by the corresponding −log10p value under each of the three categories in descending order; the top 10 GO terms were plotted as the top 30 GO enrichment analysis results (Additional file 5: Table S5 and Additional file 6: Table S6). GO analysis of the differentially expressed contiguous genes of the lncRNAs revealed (Fig. 4A) that the most significantly enriched biological processes (BPs) were complement activation, regulation of synaptic vesicle initiation, synaptic vesicle docking, chemical in vivo homeostasis, complement binding, and peptide antigen binding. The most significantly enriched cellular components (CCs) were mitochondrial ribosomes, mitochondrial large ribosomal subunits, and translation release factor complexes. The most significant molecular functions (MFs) were complement binding, peptide antigen binding, and translation release factor activity, among others. GO terms of differentially expressed mRNAs (Fig. 4B), including immune system processes, inflammatory response, and involvement in the outer side of the constituent plasma membrane, were significantly enriched.

GO enrichment analysis results. The horizontal axis is the GO entry name, and the vertical axis is −log10 P value
KEGG pathway analysis identified differentially expressed lncRNA genes (Fig. 5A and Additional file 7: Table S7), which were mainly associated with antigen processing and presentation signalling pathways, graft-versus-host disease, and cell adhesion molecules. Analysis of the differentially expressed mRNAs revealed cytokine‒cytokine receptor interactions, the NOD-like receptor signalling pathway and phagosomes (Fig. 5B and Additional file 8: Table S8).

KEGG enrichment top20 bubble map. The horizontal axis enrichment score is the enrichment score, the larger the bubble the more differential genes are contained in the entry, the bubble color changes from purple–blue–green–red; the smaller the enrichment P value, the greater the significance
LncRNA and mRNA coexpression and protein interaction network analysis
To explore the relationships between differentially expressed lncRNAs and differentially expressed mRNAs, we performed lncRNA‒mRNA coexpression analyses and constructed lncRNA‒mRNA network maps (Fig. 6A, B, Additional file 9: Table S9 and Additional file 10: Table S10). The PPI network of the top 50 DE genes is shown in the STRING database, and the interaction scores between them were all greater than 0.999 (Fig. 6C and Additional file 11: Table S11).

Coexpression circos plot (A) The outermost circle is the schematic of the autosomal distribution of the species; the second and third circles are the distribution of differentially expressed genes on the chromosome, red lines indicate upregulation, green lines indicate downregulation. The higher the bar, the higher the number of differential genes in the region; the fourth and fifth circles are the distribution of differentially expressed lncRNA on the chromosome, expressed in the same form as gene. The internal connecting lines indicate the correspondence between the Top500 coexpressed lncRNA and gene. lncRNA–miRNA network diagram (B): lncRNA are circles, miRNAs are triangles, larger graphs indicate more nodes connected to them. PPI network diagram (C): red indicates upregulated differentially expressed genes and blue indicates downregulated differentially expressed genes; the more associated genes, the larger the gene spots
Target gene network analysis and qPCR validation
The STRING database was used to predict protein‒protein interactions and map the PPI network of six lncRNAs (Additional file 12: Table S12). The results showed that H19, F630028O10Rik, Lockd, AI662270, AU020206, and Mexis were significantly associated with the target genes (Fig. 7A).

Validation of differential expression of lncRNA and target gene network analysis. The network diagram between lncRNA and target genes (A): triangles represent lncRNA and loops represent target genes. The size of the node is determined by the degree of the node, green–yellow–red indicates that log2FC is getting bigger, the darker the color the bigger the difference. qPCR validation graph (B): control and test groups were compared by the qPCR method for H19, F630028O10Rik, Lockd, AI662270, AU020206, and Mexis. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and **** P ≤ 0.0001
To ensure the accuracy and reliability of the RNA-seq data, we performed RT‒PCR to validate the expression levels of the six lncRNAs. As shown in Fig. 7B, the qPCR results of the six differentially expressed genes were consistent with the sequencing results.
Dynamic relative expression of DE lncRNAs in the cerebral tissue of mice infected with A. cantonensis
During the period of A. cantonensis infection, H19, F630028O10Rik, Lockd, AI662270, AU020206 and Mexis showed dynamic fluctuations in the mouse model compared with the negative control (Fig. 8).

Dynamic relative expression of H19, F630028O10Rik, Lockd, AI662270, AU020206, and Mexis of the cerebrum tissue in the mice model infected with A. cantonensis. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and **** P ≤ 0.0001
In the models, the expression of these six genes was significantly upregulated at 14 and 21 days after infection with A. cantonensis compared with that in the control group at the same age, and the expression level was greatest on day 14. There was no significant difference in the expression of H19, Lockd, or Mexis between 14 and 21 days. The expression levels of the other three genes decreased significantly after 21 days of infection with A. cantonensis compared with 14 days.
