
Abstract
Background
Astaxanthin is one of the strongest antioxidants in nature and has been widely used in aquaculture, food, cosmetic and pharmaceutical industries. Numerous stresses caused in the process of a large scale-culture, such as high acetate concentration, high osmolarity, high level of reactive oxygen species, high glucose concentration and acid environment, etc., limit cell growth to reach the real high cell density, thereby affecting astaxanthin production.
Results
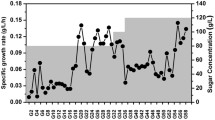
We developed an adaptive laboratory evolution (ALE) strategy to enhance the production of chemicals by improving strain tolerance against industrial fermentation conditions. This ALE strategy resulted in 18.5% and 53.7% increases in cell growth and astaxanthin production in fed-batch fermentation, respectively. Whole-genome resequencing showed that 65 mutations with amino acid substitution were identified in 61 genes of the shuffled strain Escherichia coli AST-4AS. CRISPR interference (CRISPRi) and activation (CRISPRa) revealed that the shuffled strain with higher astaxanthin production may be associated with the mutations of some stress response protein genes, some fatty acid biosynthetic genes and rppH. Repression of yadC, ygfI and rcsC, activation of rnb, envZ and recC further improved the production of astaxanthin in the shuffled strain E. coli AST-4AS. Simultaneous deletion of yadC and overexpression of rnb increased the production of astaxanthin by 32% in the shuffled strain E. coli AST-4AS.
Conclusion
This ALE strategy will be powerful in engineering microorganisms for the high-level production of chemicals.
Similar content being viewed by others

Background
Astaxanthin (3,3′-dihydroxy-β-carotene-4,4′-dione), a red-color carotenoid, is one of the strongest antioxidants in nature [1], exhibiting tremendous potential for applications in healthcare and pharmaceuticals due to its antioxidant [2], anti-inflammatory [3] and anti-cancer activity [4]. It has been widely used in aquaculture, food, cosmetic and pharmaceutical industries [5]. According to the analysis of Stratistics Market Research Consulting (MRC), the global astaxanthin market is accounted for $695.94 million in 2020 and is expected to reach $1356.47 million by 2028 growing at a compound annual growth rate of 8.7% [6]. Currently, commercial astaxanthin production relies on two routes, chemical synthesis and extraction from nature producers (such as Haematococcus pluvialis algae) [6]. However, the former route of production is not sustainable and poses concerns in health and food safety, while the latter is very costly (> $7000 per kg) [7].
To lower the cost, many studies focused on the production of astaxanthin from an inexpensive carbon source using microbes. Escherichia coli [8,9,10,11,12,13,14,15,16,17], Saccharomyces cerevisiae [18,19,20,Library screening Because astaxanthin but not the other carotenoids can efficiently diffuse through the cell membranes [41], a colorimetry of culture media (measured at 515 nm) was developed to be used for the screening of high astaxanthin-producing strains [15]. Cells from the library were diluted and then spread on LB plates. Single colonies with red color were inoculated in individual wells of a 48 deep-well microplate (4.6 mL) containing 1 mL of LB medium and incubated at 30 °C and 1000 rpm for 48 h on an MBR-420FL shaker (TAITEC, Japan). Two hundred microliter of the bacterial culture were centrifugated at 5000×g for 10 min. Then, the supernatant was transferred into a 96-well plate in which the OD515 was read using a SynergyNeo2 multi-mode reader (SynergyNeo2, BioTek, USA). Total genomic DNA of E. coli E. coli AST-4AS was extracted from mid-log phase bacterial cultures according to the manufacturer’s protocol using the TIANamp Bacterial DNA Kit (Tiangen Biotech Co., Bei**g, China). Genomic library construction and whole-genome resequencing were performed on the Illumina NOVAseq platform by Sangon Biotech (Shanghai, China). The paired-end reads from E. coli E. coli AST-4AS were aligned to the reference genome of E. coli MG1655 using BWA software (Burrows-Wheeler Aligner, version 0.7.17). Mutations, including base substitutions, deletions, and insertions, were detected by SAMtools (version 1.9), MarkDuplicates (version 4.1.1.0), and BEDTools (version 2.28.0). DNA frameshift mutations were further validated by PCR and sequencing. Mutant genes with CRISPR activation and interference were performed as described by Niu et al. [38]. The pTargetA-X series used in targeted single-gene activation or repression, with N20 sequences targeting gene loci of interest, was obtained by inverse PCR from pTargetA using primers targeting N20F/N20 genes, then cut with KpnI and self-ligated. For activation, the N20 sequence was designed to target the non-template strand upstream of the promoter. For repression, the N20 sequence was designed to target the 5′ end (about 100 bp downstream of ATG) of the gene on the non-template DNA strand. As the scRNA fragment was flanked by BamHI and BglII, the scRNA expressing vector pTargetA-XY could be reassembled from pTargetA-X and pTargetA-Y using the standard BglBrick assembly approach. To investigate the effects of activation and repression on growth and astaxanthin production, pBbB2K-dCas9*-MCPSoxS and pTargetA-X were co-transferred into E. coli AST-4. A single colony was grown in 5 mL LB (with 1% glucose) in a falcon tube at 37 °C overnights. The overnight cultures were inoculated into 10 mL of the fermentation medium at 37 °C until the OD600 reached 0.8, anhydrous tetracycline was added to the media at a final concentration of 200 nM to induce the expression of dcas9*, and then further cultured for 72 h. A single colony was inoculated into 5 mL of LB medium in a falcon tube, which was incubated overnight at 30 °C. The overnight seed culture was then inoculated into 50 mL of SBMSN medium with an initial OD600 of 0.1. SBMSN medium (pH 7.0) contained (per liter) 12 g peptone, 24 g yeast extract, 1.7 g KH2PO4, 11.42 g K2HPO4, 1 g MgCl2⋅6H2O, 1.42 g ammonium oxalate, and 2 g Tween-80. The cultures were incubated at 30 °C for 48 h in a rotary shaking incubator set to 150 rpm. Cell growth was measured according to the OD600 and converted into DCW (g/L) using a standard curve. Fed-batch fermentation was performed in a 2 L fermenter (MiniBox 2 L*2 Parallel Bioreactor System, T&J Bioengineering (Shanghai) Co. LTD, Shanghai, China) containing 1.2 L of SBMSN medium with an initial OD600 of approximately 0.1. The temperature was controlled at 30 °C, and the pH value was maintained at 7.0 by the automatic addition of NH4OH. The airflow rate was 2 L/min. Dissolved oxygen was kept above 25% by adjusting the agitation rate from 400 to 1200 rpm. A feed solution (pH 7.0) containing (per liter) 500 g glucose, 15 g peptone, 30 g yeast extract and 30 g MgSO4·7H2O was fed continuously to the fermenter using a pH–stat feeding strategy. Once the glucose was depleted, the pH rose rapidly. When the pH was greater than 7.1, the feed was automatically added to the fermenter. Samples were periodically withdrawn, and these parameters (OD600 and astaxanthin concentrations) were determined. The total RNA from E. coli cells grown in the fermentation medium for 36 h in shake flasks was isolated using an RNA extraction kit (Dongsheng Biotech, Guangzhou, China) following the manufacturer’s instructions. The first-strand cDNA was synthesized using an All-in-One™ First-Strand cDNA Synthesis kit (GeneCopoeia, Guangzhou, China). Quantitative real-time PCR was performed with the All-in-One™ qPCR Mix kit (GeneCopoeia) by an iCycler iQ5 Real Time PCR system (Bio-Rad Laboratories, California, USA). One hundred ng of cDNA was added as a template. The PCR program was set as follows: 95 °C for 10 min, followed by 45 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s, and extension at 72 °C for 15 s. The expression levels were analyzed by the 2−ΔΔCt method described by Livak et al. [42] and normalized by cysG gene expression as reference. Three biological replicates for each sample were used for qRT-PCR analysis, and three technical replicates were analyzed for each biological replicate. Gene knockout or replacement was performed according to the CRISPR-Cas method as previously described [17, 34, 43, 44]. The sgRNA plasmid pTargetB-yadC for replacement was obtained as described above for the CRISPRi system. The rnb was amplified from E. coli by PCR and then inserted into KpnI/ApaI-digested pZSKBP to obtain pZSKBP-rnb. The upstream and downstream homology arms of the yadC were amplified and then successively inserted into the MluI/AvrII and ApaI/SmaI of pZSKBP-rnb. The targeting fragment was cut from the above plasmid using MluI/SmaI and then transferred into the electrocompetent cells harboring pCas* and pTargetB-yadC to replace the corresponding gene. Cells were extracted with acetone to isolate carotenoids as previously described [9, 17]. E. coli cultures (250 μL) were harvested by centrifugation at 13,523×g for 5 min. The cell pellet was washed with water and extracted with 1 mL of acetone at 55 °C for 15 min with intermittent vortexing. The acetone supernatant after centrifugation was transferred to a new tube. Carotenoids were analyzed by HPLC (Shimadzu HPLC system, Model LC-20A, Shimadzu, Japan) using an Inertsil ODS-SP column (5 μm, 4.6 × 150 mm, GL Sciences Inc., Tokyo, Japan). The mobile phase was acetonitrile–methanol (65:35 v/v) at a flow rate of 1 mL/min. The absorbance of carotenoids at 477 nm was detected using a photodiode array detector (SPD-M20A). Carotenoid compounds were identified based on their retention times relative to standard compounds (Sigma-Aldrich, St. Louis, MO, USA). Astaxanthin was quantified by comparing the integrated peak areas with those of authentic standards. All experiments were conducted in triplicate, and data were averaged and presented as the means ± standard deviation. Oneway analysis of variance followed by Tukey’s test was used to determine significant differences using the OriginPro (version 9.0) package. Statistical significance was defined as p < 0.05.Whole-genome resequencing and data analysis
CRISPRa and CRISPRi of the mutant genes
Astaxanthin production
Quantitative real-time PCR (qRT-PCR)
Replacement of gene
Extraction and quantification of carotenoids
Statistical analysis
Availability of data and materials
Not applicable.
References
Ajikumar PK, Tyo K, Carlsen S, Mucha O, Phon TH, Stephanopoulos G. Terpenoids: opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol Pharm. 2008;5(2):167–90.
Hama S, Takahashi K, Inai Y, Shiota K, Sakamoto R, Yamada A, et al. Protective effects of topical application of a poorly soluble antioxidant astaxanthin liposomal formulation on ultraviolet-induced skin damage. J Pharm Sci-Us. 2012;101(8):2909–16.
Bennedsen M, Wang X, Willen R, Wadstrom T, Andersen LP. Treatment of H-pylori infected mice with antioxidant astaxanthin reduces gastric inflammation, bacterial load and modulates cytokine release by splenocytes. Immunol Lett. 1999;70(3):185–9.
Chew BP, Park JS, Wong MW, Wong TS. A comparison of the anticancer activities of dietary beta-carotene, canthaxanthin and astaxanthin in mice in vivo. Anticancer Res. 1999;19(3a):1849–53.
Ambati RR, Phang SM, Ravi S, Aswathanarayana RG. Astaxanthin: sources, extraction, stability, biological activities and its commercial applications—a review. Mar Drugs. 2014;12(1):128–52.
MRC. Astaxantin-Global market outlook (2020–2028). https://www.marketresearch.com/Stratistics-Market-Research-Consulting-v4058/Astaxanthin-Global-Outlook-14737524/.
Li J, Zhu DL, Niu JF, Shen SD, Wang GC. An economic assessment of astaxanthin production by large scale cultivation of Haematococcus pluvialis. Biotechnol Adv. 2011;29(6):568–74.
Lemuth K, Steuer K, Albermann C. Engineering of a plasmid-free Escherichia coli strain for improved in vivo biosynthesis of astaxanthin. Microb Cell Fact. 2011;10:29.
Lu Q, Bu YF, Liu JZ. Metabolic engineering of Escherichia coli for producing astaxanthin as the predominant carotenoid. Mar Drugs. 2017;15(10):296.
Ma T, Zhou YJ, Li XW, Zhu FY, Cheng YB, Liu Y, et al. Genome mining of astaxanthin biosynthetic genes from Sphingomonas sp. ATCC 55669 for heterologous overproduction in Escherichia coli. Biotechnol J. 2016;11(2):228–37.
Park SY, Binkley RM, Kim WJ, Lee MH, Lee SY. Metabolic engineering of Escherichia coli for high-level astaxanthin production with high productivity. Metab Eng. 2018;49:105–15.
Scaife MA, Burja AM, Wright PC. Characterization of cyanobacterial beta-carotene ketolase and hydroxylase genes in Escherichia coli, and their application for astaxanthin biosynthesis. Biotechnol Bioeng. 2009;103(5):944–55.
Wang CW, Oh MK, Liao JC. Engineered isoprenoid pathway enhances astaxanthin production in Escherichia coli. Biotechnol Bioeng. 1999;62(2):235–41.
Ye LJ, Zhu XN, Wu T, Wang W, Zhao DD, Bi CH, et al. Optimizing the localization of astaxanthin enzymes for improved productivity. Biotechnol Biofuels. 2018;11:278.
Zhang CQ, Seow VY, Chen XX, Too HP. Multidimensional heuristic process for high-yield production of astaxanthin and fragrance molecules in Escherichia coli. Nat Commun. 2018;9:1858.
Gong ZK, Wang HL, Tang JL, Bi CH, Li QY, Zhang XL. Coordinated expression of astaxanthin biosynthesis genes for improved astaxanthin production in Escherichia coli. J Agric Food Chem. 2020;68(50):14917–27.
Lu Q, Liu JZ. Enhanced astaxanthin production in Escherichia coli via morphology and oxidative stress engineering. J Agric Food Chem. 2019;67(42):11703–9.
** J, Wang Y, Yao MD, Gu XL, Li B, Liu H, et al. Astaxanthin overproduction in yeast by strain engineering and new gene target uncovering. Biotechnol Biofuels. 2018;11:230.
Lin YJ, Chang JJ, Lin HY, Thia C, Kao Y, Huang CC, et al. Metabolic engineering a yeast to produce astaxanthin. Bioresour Technol. 2017;245:899–905.
Ukibe K, Hashida K, Yoshida N, Takagi H. Metabolic engineering of Saccharomyces cerevisiae for astaxanthin production and oxidative stress tolerance. Appl Environ Microb. 2009;75(22):7205–11.
Wang RZ, Gu XL, Yao MD, Pan CH, Liu H, **ao WH, et al. Engineering of beta-carotene hydroxylase and ketolase for astaxanthin overproduction in Saccharomyces cerevisiae. Front Chem Sci Eng. 2017;11(1):89–99.
Zhou PP, Ye LD, **e WP, Lv XM, Yu HW. Highly efficient biosynthesis of astaxanthin in Saccharomyces cerevisiae by integration and tuning of algal crtZ and bkt. Appl Microbiol Biotechnol. 2015;99(20):8419–28.
Zhou PP, **e WP, Li AP, Wang F, Yao Z, Bian Q, et al. Alleviation of metabolic bottleneck by combinatorial engineering enhanced astaxanthin synthesis in Saccharomyces cerevisiae. Enzyme Microb Technol. 2017;100:28–36.
Zhou PP, Li M, Shen B, Yao Z, Bian Q, Ye LD, et al. Directed coevolution of beta-carotene ketolase and hydroxylase and its application in temperature-regulated biosynthesis of astaxanthin. J Agric Food Chem. 2019;67(4):1072–80.
Henke NA, Heider SAE, Peters-Wendisch P, Wendisch VF. Production of the marine carotenoid astaxanthin by metabolically engineered Corynebacterium glutamicum. Mar Drugs. 2016;14(7):124.
Kildegaard KR, Adiego-Perez B, Belda DD, Khangura JK, Holkenbrink C, Borodina I. Engineering of Yarrowia lipolytica for production of astaxanthin. Syn Syst Biotechnol. 2017;2(4):287–94.
Chen YY, Shen HJ, Cui YY, Chen SG, Weng ZM, Zhao M, et al. Chromosomal evolution of Escherichia coli for the efficient production of lycopene. BMC Biotechnol. 2013;13:6.
Zhao J, Li QY, Sun T, Zhu XN, Xu HT, Tang JL, et al. Engineering central metabolic modules of Escherichia coli for improving beta-carotene production. Metab Eng. 2013;17:42–50.
Shen HJ, Cheng BY, Zhang YM, Tang L, Li Z, Bu YF, et al. Dynamic control of the mevalonate pathway expression for improved zeaxanthin production in Escherichia coli and comparative proteome analysis. Metab Eng. 2016;38:180–90.
Fraser PD, Sandmann G. In vitro assays of three carotenogenic membrane-bound enzymes from Escherichia coli transformed with different crt genes. Biochem Biophys Res Commun. 1992;185(1):9–15.
Lennen RM, Herrgard MJ. Combinatorial strategies for improving multiple-stress resistance in industrially relevant Escherichia coli strains. Appl Environ Microb. 2014;80(19):6223–42.
Gong ZW, Nielsen J, Zhou YJJ. Engineering robustness of microbial cell factories. Biotechnol J. 2017;12(10):1700014.
Jiang T, Li CY, Teng YX, Zhang RH, Yan YJ. Recent advances in improving metabolic robustness of microbial cell factories. Curr Opin Biotechnol. 2020;66:69–77.
Niu FX, He X, Wu YQ, Liu JZ. Enhancing production of pinene in Escherichia coli by using a combination of tolerance, evolution, and modular co-culture engineering. Front Microbiol. 2018;9:1623.
Niu FX, He X, Huang YB, Liu JZ. Biosensor-guided atmospheric and room-temperature plasma mutagenesis and shuffling for high-level production of shikimic acid from sucrose in Escherichia coli. J Agric Food Chem. 2020;68(42):11765–73.
Ye LD, Zhao H, Li Z, Wu JC. Improved acid tolerance of Lactobacillus pentosus by error-prone whole genome amplification. Bioresour Technol. 2013;135:459–63.
He XT, Xue TL, Ma YY, Zhang JY, Wang ZQ, Hong JF, et al. Identification of functional butanol-tolerant genes from Escherichia coli mutants derived from error-prone PCR-based whole-genome shuffling. Biotechnol Biofuels. 2019;12:73.
Niu FX, Huang YB, Ji LN, Liu JZ. Genomic and transcriptional changes in response to pinene tolerance and overproduction in evolved Escherichia coli. Syn Syst Biotechnol. 2019;4(3):113–9.
Aguilar C, Escalante A, Flores N, de Anda R, Riveros-McKay F, Gosset G, et al. Genetic changes during a laboratory adaptive evolution process that allowed fast growth in glucose to an Escherichia coli strain lacking the major glucose transport system. BMC Genomics. 2012;13:385.
Datta S, Costantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379:109–15.
Kidd P. Astaxanthin, cell membrane nutrient with diverse clinical benefits and anti-aging potential. Altern Med Rev. 2011;16(4):355–64.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25(4):402–8.
Jiang Y, Chen B, Duan CL, Sun BB, Yang JJ, Yang S. Multigene editing in the Escherichia coli genome via the crispr-cas9 system. Appl Environ Microb. 2015;81(7):2506–14.
Shen YP, Liao YL, Lu Q, He X, Yan ZB, Liu JZ. ATP and NADPH engineering of Escherichia coli to improve the production of 4-hydroxyphenylacetic acid using CRISPRi. Biotechnol Biofuels. 2021;14(1):100.
Acknowledgements
Not applicable.
Funding
This work was funded by National Key R&D Program of China (2020YFA0906900), National Natural Science Foundation of China (Grant NO. 31901024 and 32071422), Guangdong Basic and Applied Basic Research Foundation (NO. 2019B1515210006), Key-Area Research and Development Program of Guangdong Province (NO. 2020B020226007), the Open Fund Project of Shenzhen Innovation Institute of Synthetic Biology (DWKF20190003), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (20lgpy113).
Author information
Authors and Affiliations
Contributions
QL performed all of the experimental works. XLZ constructed the sgRNA plasmids. JZL designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The colorimetry of culture media for high throughput screening. Fig. S2. Production of astaxanthin determined using OD515 by the evolved strains after ARTP mutation. Fig. S3. Production of astaxanthin determined using OD515 by the strains after error-prone whole-genome shuffling. Fig. S4. HPLC analysis of carotenoid products extracted from E. coli AST-4AS cultured in 2-L bioreactor. Fig. S5. Effect of CRISPR repressing of the mutated gene on the astaxanthin production. Table S1. Mutated genes identified in E. coli AST-4AS. Table S2. Primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Q., Zhou, XL. & Liu, JZ. Adaptive laboratory evolution and shuffling of Escherichia coli to enhance its tolerance and production of astaxanthin. Biotechnol Biofuels 15, 17 (2022). https://doi.org/10.1186/s13068-022-02118-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-022-02118-w