
Abstract
Background
Diesel exhaust (DE) induces neutrophilia and lymphocytosis in experimentally exposed humans. These responses occur in parallel to nuclear migration of NF-κB and c-Jun, activation of mitogen activated protein kinases and increased production of inflammatory mediators. There remains uncertainty regarding the impact of DE on endogenous antioxidant and xenobiotic defences, mediated by nuclear factor erythroid 2-related factor 2 (Nrf2) and the aryl hydrocarbon receptor (AhR) respectively, and the extent to which cellular antioxidant adaptations protect against the adverse effects of DE.
Methods
Using immunohistochemistry we investigated the nuclear localization of Nrf2 and AhR in the epithelium of endobronchial mucosal biopsies from healthy subjects six-hours post exposure to DE (PM10, 300 µg/m3) versus post-filtered air in a randomized double blind study, as a marker of activation. Cytoplasmic expression of cytochrome P450s, family 1, subfamily A, polypeptide 1 (CYP1A1) and subfamily B, Polypeptide 1 (CYP1B1) were examined to confirm AhR activation; with the expression of aldo–keto reductases (AKR1A1, AKR1C1 and AKR1C3), epoxide hydrolase and NAD(P)H dehydrogenase quinone 1 (NQO1) also quantified. Inflammatory and oxidative stress markers were examined to contextualize the responses observed.
Results
DE exposure caused an influx of neutrophils to the bronchial airway surface (p = 0.013), as well as increased bronchial submucosal neutrophil (p < 0.001), lymphocyte (p = 0.007) and mast cell (p = 0.002) numbers. In addition, DE exposure enhanced the nuclear translocation of the AhR and increased the CYP1A1 expression in the bronchial epithelium (p = 0.001 and p = 0.028, respectively). Nuclear translocation of AhR was also increased in the submucosal leukocytes (p < 0.001). Epithelial nuclear AhR expression was negatively associated with bronchial submucosal CD3 numbers post DE (r = −0.706, p = 0.002). In contrast, DE did not increase nuclear translocation of Nrf2 and was associated with decreased NQO1 in bronchial epithelial cells (p = 0.02), without affecting CYP1B1, aldo–keto reductases, or epoxide hydrolase protein expression.
Conclusion
These in vivo human data confirm earlier cell and animal-based observations of the induction of the AhR and CYP1A1 by diesel exhaust. The induction of phase I xenobiotic response occurred in the absence of the induction of antioxidant or phase II xenobiotic defences at the investigated time point 6 h post-exposures. This suggests DE-associated compounds, such as polycyclic aromatic hydrocarbons (PAHs), may induce acute inflammation and alter detoxification enzymes without concomitant protective cellular adaptations in human airways.
Similar content being viewed by others


Introduction
Traffic-related particulate matter, to which diesel exhaust (DE) is a significant contributor, is associated with increased cardiorespiratory morbidity [1,2,3]. Many studies have shown that DE exposure triggers acute neutrophilic and lymphocytic inflammation, characterized by the release of a variety of chemokines and cytokines, CXCL8 (IL-8), CXCL1 (Gro-alpha) members of CXC chemokines and Th2 cytokine (IL-13), along with inflammatory cell recruitment to the conducting airways [4,5,6]. Numerous reports have suggested that the generation of reactive oxidative species (ROS) and the subsequent oxidative stress induced by inhaled particulate matter (PM) and PM associated chemicals, is a key trigger for the observed inflammation [4, 2 panel B). Diesel exhaust comprise both particulate components and gases, mainly oxides of nitrogen, including NO2. We do not suggest NO2 to have influenced the epithelial and submucosal inflammatory events by DE. We have carried out studies with NO2 at a higher concentration (2 ppm for 4 h) than employed here, without any bronchial mucosal inflammatory responses [33]. Previous studies have shown that fresh DE particles have negligible inherent oxidative potential, compared to ambient PM. For example, determination of oxidative potential (OP) of DEPs (PM 0.1–10) generated under idling engine condition and European Transient Cycle (ETC) urban engine condition was assessed, and demonstrated that oxidizing capacity of DEPs under idling condition was higher than during ETC urban condition. The oxidative properties observed from both these experimental tail-pipe emissions were minor compared to ambient PM [34]. Barath et al. [35] reported that fresh DE particles were abundant in organic species, comparing PAHs from the idling and transient running conditions. The former was shown to have four times higher concentrations of total PAHs in the exposure chamber, mainly related to higher idling gaseous PAH concentrations. PM-associated PAH concentrations were similar under both running conditions, but with differences in profile. PM-associated PAHs including phenanthrene, fluoranthene and pyrene were present in higher concentrations under transient load and speed. However, under idling condition, PM-associated PAHs distribution included heavier PAH (4-rings) species [34]. These PAHs present in the DE are known to be exogenous AhR ligands. In the current study, the epithelial nuclear translocated p–c-Jun expression was positively associated with nuclear translocated/activated Nrf2 at post filtered air exposure, while DE exposure altered this association (Fig. 1E, F). This suggests that higher or lower redox sensitive activity may demand higher/lower Nrf2 activity to keep the antioxidant defense in balance. Decreased NQO1 expression together with absence of Nrf2 activation in this study following DE exposure could potentially be aggravated in more susceptible individuals, such as asthmatic and COPD subjects, with preexisting oxidative stress and/or impaired antioxidant defences [35, 36].
Conclusion
Six-hours after an acute high-dose diesel challenge, in the presence of a robust central airway neutrophilia and lymphocytosis, we observed evidence of AhR nuclear translocation/activation, associated with increased CYP1A1 expression. This occurred in the absence of Nrf2 activation/nuclear translocation. The present data therefore suggest that the acute effects of diesel exhaust in humans are likely driven by AhR ligands within the organic fraction, activating the AhR. The c-Jun activity as a redox sensitive molecule, in the absence of activated Nrf2 and inflammation, may be secondary to this, as indicated from previous observations in airway nerves [37].
Due to the invasive nature of human studies that employ bronchoscopy, there are limitations when it comes to the number of samples that can be obtained. Therefore, the present study is limited in that it only allows a brief snap-shot in time of multiple overlap** molecular activity. There is clearly a need to obtain a better kinetic understanding of the early induction of these transcription factors and their interaction, but what the present study does imply is that the simple schema, in which inhaled combustion particles first induce oxidative stress in the lung, triggering downstream responses, ranging from adaptation to cell death, does always not seem to be operating and that the AhR, is suggested to play a central role in the early airway response to diesel exhaust. Given our previous observations of the induction of NFκB (p65) and AP-1 (phosphorylated c-jun) at this early time point [11], more work is required to understand how these transcription factors interact with the AhR in the early response, as well as the extent to which the induction of phase I xenobiotic metabolism may result in transient changes in intracellular ROS generation following diesel challenge. However, the role of the AhR in mucosal defences is complex and in interrogating its responses to exogenous ligands, there is also a need to consider whether pathways generating endogenous AhR ligands, such as via induction of indoleamine 2,3-dioxygenase (IDO1), may be playing a role. The evidence presented here suggests that the response of the AhR to exogeous ligands within diesel exhaust is associated with the acute inflammatory response observed.
Material and methods
Subjects
Sixteen healthy volunteers (9 females and 7 males), with a mean age of 24 years (range 20–31), were enrolled in the study. All subjects were non-smokers with normal lung function and negative skin prick tests against a standard panel of airborne allergens – see Additional file 4: Table S3. They were all free from respiratory tract infections at least six weeks prior to, and during, the study period. The study was approved by the local Ethical Review Board at Umeå University and performed in accordance with the Declaration of Helsinki, with written informed consent of all participating volunteers.
DE exposure
All subjects were exposed on two different occasions, once to filtered air and once to diesel engine exhaust, in a randomized order, at least three weeks apart. Each exposure lasted for one hour, during which the subjects alternated between fifteen-minute intervals of rest and exercise on a bicycle ergometer, with the workload adjusted to achieve a minute ventilation of 20 L/min/m2 body surface. Diesel exhaust was generated by an idling Volvo diesel engine Volvo (TD45, 4.5 L, 4 Cylinders, 1991, 680 rpm) running on Gasoil E10 (Preem, Sweden). The majority of the exhaust was shunted away, while the remaining part was diluted with filtered air and fed into the exposure chamber where air pollution parameters were continuously monitored. The mean concentration of particulates with a mass median diameter smaller than 10 µm (PM10) was 290 ± 27 µg/m3 during the diesel exhaust exposures. This was associated with concentrations of nitric oxide (NO) of 2.9 ± 0.37 ppm, nitrogen dioxide (NO2) of 0.84 ± 0.10 ppm, total hydrocarbons (HC) of 1.2 ± 0.15 ppm and carbon monoxide (CO) of 2.4 ± 0.53 ppm. Chemical characterization of the exposure emissions has been published elsewhere [34, 38].
Bronchoscopy and processing of samples
Bronchoscopy was performed six hours after both exposures using a flexible video bronchoscope (Olympus BF IT160, Tokyo, Japan). Endobronchial mucosal biopsies were taken either from the anterior aspect of the main carina and the subcarinae of the 3rd and 4th generation airways of the right side or from the posterior aspect of the main carina and the corresponding subcarinae on the left side. Bronchial wash (BW, 2 × 20 ml) and bronchoalveolar lavage (BAL, 3 × 60 ml) with saline were carried out on the contra-lateral side, in a pre-determined randomized way. The aspirates recovered from the first and second 20 ml instillations of the BW and the pooled BAL were collected into separate siliconized containers placed on ice. All lavage samples were filtered through a nylon filter (pore diameter 100 µm) and centrifuged at 400 g for 15 min. Cell pellets were re-suspended in PBS at a cell concentration of 106 cells/ml. Differential cell counts were performed on slides made by cyto-centrifuge preparation and stained with May-Grünwald Giemsa and 400 cells per slide were counted. Biopsies were fixed overnight (16–20 h) at −20 °C in chilled acetone, containing protease inhibitors (20 mM iodoacetamide and 2 mM phenylmethylsulphonyl fluoride). After fixation the biopsies were processed into glycolmethacrylate (GMA) resins, as described earlier [39].
Immunohistochemistry
Antibodies for detection of inflammatory cells, including neutrophils and mast cells, were purchased from Dako (Glostrup, Denmark). Human anti CD3, which is a T cell marker, and human anti ECP (EG2), an eosinophil marker, were purchased from Serotec (Oxford, UK) and Diagnostic development (Uppsala, Sweden), respectively. Antibodies used to detect transcription factors and detoxification enzymes, including p–c-Jun, NQO1, c-Fos (mouse monoclonal antibodies), Nrf2 and AHR (rabbit polyclonal antibodies), were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The supplier for the antibodies against Cyp1A1, Cyp1B1, EPHX (mouse monoclonal antibodies) and Aldo–keto reductases (AKR1A1, AKR1C1 and AKR1C3) was Abcam (Cambridge, UK). Biotinylated rabbit anti-mouse and swine anti-rabbit antibodies were purchased from Dako.
The staining procedure has been previously described in detail [4, 11]. Briefly, the GMA-embedded biopsies were cut in 2-µm thin slices and floated onto ammonia water (1:500). They were collected onto 0.01% poly-L-lysine-coated glass slides and dried at room temperature for one hour. For staining of inflammatory cells, the sections were treated to block endogenous peroxidases and nonspecific antibody binding, and the primary antibody was applied and incubated at room temperature overnight. After rinsing, biotinylated rabbit anti-mouse antibodies against the monoclonal antibodies, and swine anti-rabbit antibodies against polyclonal antibodies (Dako), were applied for two hours, followed by VECTASTAIN Elite ABC kit (Vector Laboratories, Burlingame, USA) for another two hours. Sections for submucosal analysis were developed with aminoethyl carbazole (AEC) as substrate. All sections were counterstained with Mayer´s hematoxylin. Sections where the primary antibody was omitted served as negative controls. The staining procedure was partially modified for transcription factors and detoxification enzymes. Supplementary steps were added in order to increase the permeability of the cells. Sections for epithelial measurements were developed as a brown colour with 3,3-diaminobenzidine (DAB). A detailed description of the staining procedure has previously been published [11].
Quantification of the immunohistochemical staining and other analyses were performed by one person who was unaware of how the coded samples corresponding to the exposures. All analyses were done double blinded, with codes broken only after completion of the full analysis and statistics. When quantifying the immunohistochemical staining, a light microscope was used to count inflammatory cells according to their immunoreactivity with specific antibodies in the epithelium and the submucosa respectively, excluding mucosal glands, blood vessels and smooth muscle. A computer-assisted image analysis program (Leica Q500IW, Leica Cambridge UK) was used to calculate the length of the epithelium and the area of the submucosa. Cell counts were expressed as cells/mm2 in the selected submucosa area. The images presented in (Fig. 3), displays the immunoreactivity patterns for transcription factors and enzymes in the bronchial epithelium. Quantification of transcription factors in the bronchial epithelium, total staining (cytoplasmic plus nuclear) and enzymes, immunoreactivity was quantified using a Leica DFC 320 camera (Cambridge, UK). The camera was connected to a Leica imaging workstation, specific PC software (Leica Q500IW; Leica, Cambridge, UK). Detection of an appropriate color was quantified using binary definition of color images as displayed on the screen. The binary image required the user to define which pixel in the image was to be considered for measurement. The total staining (cytoplasmic plus nuclear) and enzymes were expressed as the percentage of the total epithelial area showing positive immunoreactivity to the antibodies. It was possible to distinguish between nuclear and cytoplasmic staining using a light microscope. Positive staining of the nucleus was expressed as the number of positive nuclei/mm2 of selected epithelium area.

Immunoreactivity within the bronchial epithelium. Upper left shows immunoreactivity to NQO1 post-air exposure and upper right shows NQO1 post-DE exposure. Middle panel shows immunoreactivity to AhR antibody, middle left, post-air exposure and middle right, post-DE exposure. Immunoreactivity to P–C-Jun antibody is shown in lower panel, with lower left at post-air exposure and lower right at post-DE exposure. Narrow arrows indicate positive intracytoplasmic staining and bold arrows indicate nuclear staining. Scale bars = 50 μm (micro meters)
Due to lack of double staining or sequential sections for co-localization, we were not able to quantify nuclear translocated AhR in specific cells in the submucosa. Therefore, AhR activation in the submucosal leukocytes was expressed as the number of nuclear translocated AhR in the leukocytes/mm2 submucosa area. Despite to the lack of double staining or sequential sections for co-localization, we could confirm, AhR leukocyte nuclear translocation and co-localization with submucosal CD3+ cells post DE exposure ( Fig. 2 panel B).
Antioxidant analysis
Cell-free BW and BAL supernatants were analysed for total protein, GSH, GSSG, vitamin C (AA and DHA), and UA concentration, as previously described [40]. Briefly, total glutathione concentrations were measured using the GSSG-reductase-DTNB recycling method. AA and UA were measured simultaneously by reverse phase HPLC with electrochemical detection, as previously described [40]. Total vitamin C (DHA + AA) was measured by pre-treating samples with 50 mM Tris(2-carboxylethyl) phosphine for 15 min to reduce DHA and then performing the lipid extraction and HPLC analysis as described above. The DHA concentration was then calculated by subtracting the AA concentration from the total vitamin C concentration.
Cytokine analyses
Cytokine concentrations were determined in untreated lavage fluids.
IL-17A, IL-17F and TGF-β1 concentrations were measured with commercial ELISA kits (Gen-Probe Diaclone, France) and IL-17E concentrations was determined by using Human IL-17E ELISA Construction Kit (Antigenix America). IL-6 and IL-10 concentrations were measured using high-sensitivity ELISA kits, R&D Systems (Abingdon United Kingdom), which the lower limit detection for IL-6 was 0.007–0.090 (mean 0.031) and for IL-10 was 0.03–0.17 (mean 0.09) for IL-10. Here a lower limit of detection for both IL-6 and IL-10 have been determined by adding 3SD of O.D. blank value. The calculated concentrations below the lower standard point, but above the sensitity to detect minimum dose, recommended in the kit, were accepted as valid values.
Statistical analysis
Subjects acted as their own controls, and comparison of post-air exposure and post-diesel exhaust exposure were performed using Wilcoxon’s nonparametric signed-rank test. A p value of 0.05, or less, was considered significant and data are presented as median and interquartile range. Correlation analyses were carried out using Spearman’s rank order correlation with a p value of 0.05, or less, was considered significant. Comparisons were performed using absolute value in a given parameter after air or diesel exposure. Comparisons of the change in a given parameter were performed using (post-diesel value minus post-air value). All statistical analyses were performed using SPSS version 27.0 (SPSS Inc., Chicago, USA). Graphical presentation of absolute value in a given parameter after air and diesel for each subject were performed using GraphPad software, prism version 9 (San Diego, CA, USA).
Availability of data and materials
All relevant data are included in the manuscript and supporting information. These are also available from the authors upon reasonable request.
References
Gauderman WJ, Avol E, Gilliland F, Vora H, Thomas D, Berhane K, et al. The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med. 2004;351(11):1057–67.
Russell AG, Brunekreef B. A focus on particulate matter and health. Environ Sci Technol. 2009;43(13):4620–5.
Mills NL, Donaldson K, Hadoke PW, Boon NA, MacNee W, Cassee FR, et al. Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med. 2009;6(1):36–44.
Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159(3):702–9.
Salvi SS, Nordenhall C, Blomberg A, Rudell B, Pourazar J, Kelly FJ, et al. Acute exposure to diesel exhaust increases IL-8 and GRO-alpha production in healthy human airways. Am J Respir Crit Care Med. 2000;161(2 Pt 1):550–7.
Pourazar J, Frew AJ, Blomberg A, Helleday R, Kelly FJ, Wilson S, et al. Diesel exhaust exposure enhances the expression of IL-13 in the bronchial epithelium of healthy subjects. Respir Med. 2004;98(9):821–5.
**a T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, et al. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environ Health Perspect. 2004;112(14):1347–58.
Behndig AF, Mudway IS, Brown JL, Stenfors N, Helleday R, Duggan ST, et al. Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respir J. 2006;27(2):359–65.
Mudway IS, Stenfors N, Duggan ST, Roxborough H, Zielinski H, Marklund SL, et al. An in vitro and in vivo investigation of the effects of diesel exhaust on human airway lining fluid antioxidants. Arch Biochem Biophys. 2004;423(1):200–12.
Kelly FJ. Oxidative stress: its role in air pollution and adverse health effects. Occup Environ Med. 2003;60(8):612–6.
Pourazar J, Mudway IS, Samet JM, Helleday R, Blomberg A, Wilson SJ, et al. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L724–30.
Pourazar J, Blomberg A, Kelly FJ, Davies DE, Wilson SJ, Holgate ST, et al. Diesel exhaust increases EGFR and phosphorylated C-terminal Tyr 1173 in the bronchial epithelium. Part Fibre Toxicol. 2008;5:8.
Baulig A, Garlatti M, Bonvallot V, Marchand A, Barouki R, Marano F, et al. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L671–9.
Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, et al. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am J Respir Cell Mol Biol. 2001;25(4):515–21.
**ao GG, Wang M, Li N, Loo JA, Nel AE. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem. 2003;278(50):50781–90.
Penning TM. Aldo-keto reductase regulation by the Nrf2 system: implications for stress response, chemotherapy drug resistance, and carcinogenesis. Chem Res Toxicol. 2017;30(1):162–76.
Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I, et al. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: biochemical, pharmacological and toxicological implications. J Proteomics. 2014;108:171–87.
Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21(1):102–16.
Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30(9):447–54.
Kohle C, Bock KW. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem Pharmacol. 2007;73(12):1853–62.
Kumar A, Dailey LA, Swedrowska M, Siow R, Mann GE, Vizcay-Barrena G, et al. Quantifying the magnitude of the oxygen artefact inherent in culturing airway cells under atmospheric oxygen versus physiological levels. FEBS Lett. 2016;590(2):258–69.
Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501(1):116–23.
Jaiswal AK. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic Biol Med. 2000;29(3–4):254–62.
Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353(6345):670–4.
Soontjens C, Holmberg K, Westerholm R, Rafter J. Characterisation of polycyclic aromatic compounds in diesel exhaust particulate extract responsible for aryl hydrocarbon receptor activity. Atmos Environ. 1997;31(2):219–25.
Misaki K, Suzuki M, Nakamura M, Handa H, Iida M, Kato T, et al. Aryl hydrocarbon receptor and estrogen receptor ligand activity of organic extracts from road dust and diesel exhaust particulates. Arch Environ Contam Toxicol. 2008;55(2):199–209.
Totlandsdal AI, Lag M, Lilleaas E, Cassee F, Schwarze P. Differential proinflammatory responses induced by diesel exhaust particles with contrasting PAH and metal content. Environ Toxicol. 2015;30(2):188–96.
Hao N, Whitelaw ML. The emerging roles of AhR in physiology and immunity. Biochem Pharmacol. 2013;86(5):561–70.
Poulain-Godefroy O, Boute M, Carrard J, Alvarez-Simon D, Tsicopoulos A, de Nadai P. The aryl hydrocarbon receptor in asthma: friend or foe? Int J Mol Sci. 2020;21(22):8797.
Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19(3):184–97.
Denison MS, Faber SC. And now for something completely different: diversity in ligand-dependent activation of Ah receptor responses. Curr Opin Toxicol. 2017;2:124–31.
Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. 2018;48(1):19–33.
Blomberg A, Krishna MT, Bocchino V, Biscione GL, Shute JK, Kelly FJ, et al. The inflammatory effects of 2 ppm NO2 on the airways of healthy subjects. Am J Respir Crit Care Med. 1997;156(2 Pt 1):418–24.
Barath S, Mills NL, Lundback M, Tornqvist H, Lucking AJ, Langrish JP, et al. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Part Fibre Toxicol. 2010;7:19.
Kelly FJ, Mudway I, Blomberg A, Frew A, Sandstrom T. Altered lung antioxidant status in patients with mild asthma. Lancet. 1999;354(9177):482–3.
Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33: 101544.
Robinson RK, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Chen S, et al. Mechanistic link between diesel exhaust particles and respiratory reflexes. J Allergy Clin Immunol. 2018;141(3):1074–84.
Lucking AJ, Lundback M, Mills NL, Faratian D, Barath SL, Pourazar J, et al. Diesel exhaust inhalation increases thrombus formation in man. Eur Heart J. 2008;29(24):3043–51.
Britten KM, Howarth PH, Roche WR. Immunohistochemistry on resin sections: a comparison of resin embedding techniques for small mucosal biopsies. Biotech Histochem. 1993;68(5):271–80.
Dove RE, Leong-Smith P, Roos-Engstrand E, Pourazar J, Shah M, Behndig AF, et al. Cigarette smoke-induced induction of antioxidant enzyme activities in airway leukocytes is absent in active smokers with COPD. Eur Clin Respir J. 2015;2:27837.
Acknowledgements
IM and FJK are part funded by the National Institute for Health Research (NIHR) Health Protection Research Unit in Environmental Exposures and Health, a partnership between Public Health England and Imperial College London. The views expressed are those of the author(s) and not necessarily those of the NIHR, Public Health England or the Department of Health and Social Care.
Funding
Open access funding provided by Umea University. This work was supported by the Swedish Heart Lung Foundation, Swedish Medical Research Council, Umeå University, Västerbotten County Council (Sputspetsanslag).
Author information
Authors and Affiliations
Contributions
MF, AFB, JAB, ISM, FJK, AB, TS and JP conceived and participated in the study design. AFB, SB and AB carried out the clinical bronchoscopy studies and collected the data. MF, RD and JP were responsible for the laboratory and immunohistochemistry analyses. MF, JP, RD, ISM, DG, AB and TS took part in data analysis and interpretation. MF and JP drafted the manuscript and all authors participated in critical review of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local Ethical Review Board at Umeå University and performed in accordance with the Declaration of Helsinki, with written informed consent of all participating volunteers. Further information is given in the material and method section.
Competing interests
The authors have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
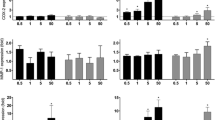
Transcription factors, enzymes and submucosal cell expression, graphically presentation of air and DE data points. Definition of abbreviations: Epi. = epithelium, nucl. = nucleus, Subm. = submucosal, leu. = leukocyte. Each data point (absolute value post-air and post-deisel for each subject) given graphically, performed using GraphPad software, prism version 9. Total staining (cytoplasmic + nucleus expression) and enzyme staining, expressed as % of the selected epithelial area. Staining of the nucleus expressed as the number of positively stained nuclei/mm2 of the selected epithelial area. Submucosal leukocyte nuclear AhR and submucosal cells are expressed as nuclei or cell numbers/mm2 submucosa area, (n = 16).
Additional file 2. Table S1.
BW cytokine expression.
Additional file 3. Table S2.
Bronchial wash antioxidant concentrations.
Additional file 4. Table S3.
Subject demographics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Friberg, M., Behndig, A.F., Bosson, J.A. et al. Human exposure to diesel exhaust induces CYP1A1 expression and AhR activation without a coordinated antioxidant response. Part Fibre Toxicol 20, 47 (2023). https://doi.org/10.1186/s12989-023-00559-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-023-00559-1