
Abstract
Background
Chronic kidney disease (CKD) is increasingly recognized as a stroke risk factor, but its exact relationship with cerebrovascular disease is not well-understood. We investigated the development of cerebral small vessel disease using in vivo and in vitro models of CKD.
Methods
CKD was produced in aged C57BL/6J mice using an adenine-induced tubulointerstitial nephritis model. We analyzed brain histology using Prussian blue staining to examine formation of cerebral microhemorrhage (CMH), the hemorrhagic component of small vessel disease and the neuropathological substrate of MRI-demonstrable cerebral microbleeds. In cell culture studies, we examined effects of serum from healthy or CKD patients and gut-derived uremic toxins on brain microvascular endothelial barrier.
Results
CKD was induced in aged C57BL/6J mice with significant increases in both serum creatinine and cystatin C levels (p < 0.0001) without elevation of systolic or diastolic blood pressure. CMH was significantly increased and positively correlated with serum creatinine level (Spearman r = 0.37, p < 0.01). Moreover, CKD significantly increased Iba-1-positive immunoreactivity by 51% (p < 0.001), induced a phenotypic switch from resting to activated microglia, and enhanced fibrinogen extravasation across the blood–brain barrier (BBB) by 34% (p < 0.05). On analysis stratified by sex, the increase in CMH number was more pronounced in male mice and this correlated with greater creatinine elevation in male compared with female mice. Microglial depletion with PLX3397 diet significantly decreased CMH formation in CKD mice without affecting serum creatinine levels. Incubation of CKD serum significantly reduced transendothelial electrical resistance (TEER) (p < 0.01) and increased sodium fluorescein permeability (p < 0.05) across the endothelial monolayer. Uremic toxins (i.e., indoxyl sulfate, p-cresyl sulfate, and trimethylamine-N-oxide) in combination with urea and lipopolysaccharide induced a marked drop in TEER compared with the control group (p < 0.0001).
Conclusions
CKD promotes the development of CMH in aged mice independent of blood pressure but directly proportional to the degree of renal impairment. These effects of CKD are likely mediated in part by microglia and are associated with BBB impairment. The latter is likely related to gut-derived bacteria-dependent toxins classically associated with CKD. Overall, these findings demonstrate an important role of CKD in the development of cerebral small vessel disease.
Similar content being viewed by others

Background
Chronic kidney disease (CKD) is a major public health issue that affects 15% of U.S. adults, leading to impaired quality of life [1]. Meta-analysis and systemic review of population-based studies provide strong evidence supporting CKD as an independent risk factor for cerebral small vessel disease and cognitive impairment [2,3,4]. Notably, end-stage renal disease is associated with a substantially higher risk of both ischemic and hemorrhagic strokes [3] as well as accelerated brain aging and cognitive decline [5], and consequently, higher morbidity and mortality [6]. Postmortem examination of CKD human brains showed an increased prevalence of cerebral small vessel disease and highlighted the frequency of microvascular calcification in those brains [7]. Pathways that promote cerebral small vessel disease in the uremic milieu include loss of calcium/phosphorus homeostasis, blood pressure variability, retention of vascular toxins, and chronic inflammation [8]. Investigations into the relationship between CKD and cerebral small vessel disease subtypes are needed to identify novel prevention and treatment strategies in CKD patients.
Cerebral microhemorrhages (CMH) are the pathological substrate for cerebral microbleeds, which represent focal hemosiderin/iron deposits on MRI and are linked to cognitive impairment and ischemic and hemorrhagic stroke. Given that cerebral microbleeds are strongly age-dependent [9] and are present in up to 50% of hemodialysis patients [42,43,44]. Microglial dynamics are important in maintaining brain homeostasis. Microglia are maintained in a resting state with a morphology characterized by long, ramified processes extending from the soma and terminating with bulbous endings; they can be activated when brain injuries are present, transforming into a phagocytic appearance, exhibiting large, rounded soma with no or few processes [25, 45]. Impaired microglial function has been implicated in aging [46] and Alzheimer’s disease [47]. CKD-induced neuroinflammation has been associated with increased microglia/macrophage recruitment, a shift from an anti-inflammatory M2 to a pro-inflammatory M1 phenotype [48], and the formation of NLRP3 inflammasomes [49]. Although morphological profiles of microglia have been well-characterized in rodents [50], little is known about their morphological changes in the context of CKD, as well as the neuropathological consequences. In the current study, CKD is associated with microglial activation, shown by increased Iba-1 immunoreactivity and a phenotypic switch from resting to activated microglia (Fig. 3A–C).
We used Iba-1 immunostaining which identifies both microglia and macrophages [48]. We characterized the morphology of microglia at higher magnification (40x) to distinguish microglia from macrophages, which allowed us to determine the contribution of microglia in CKD-induced neuroinflammation (Fig. 3F). Our prior mouse work showed a positive correlation between Iba-1 immunoreactivity and CMH number [14]. Thus, the causal relationship between microglial activation and CMH formation was investigated in the current study using PLX3397 diet allowing for microglial depletion (Fig. 4). CKD-induced CMH formation was significantly decreased in aged mice with microglial depletion (Fig. 4F), while serum creatinine levels were found not to be affected (Fig. 4B). These findings indicate that CKD-induced CMH formation is at least partly mediated by microglial activation.
The BBB is formed by endothelial cells lining the capillary wall, astrocyte end-feet surrounding the capillary, and pericytes embedded in the baseline membrane, thus creating a physical barrier between the peripheral circulation and the central nervous system. The tight junctions between the endothelial cells serve to restrict passage of blood-borne substances (e.g., fibrinogen) into the brain and play a crucial role in brain homeostasis [51]. Impaired endothelial tight junctions at the BBB are well-characterized in aging brains [52] and can lead to passage of iron into the brain [53]. We have previously shown disruption in BBB tight junction proteins in young CKD mice during an inflammatory state [13]. Following the previous observations, we examined the passage of fibrinogen into the brains of CKD mice. Fibrinogen enters the brain after BBB injury and can be converted into insoluble fibrin, contributing to neuroinflammation and neuronal damage in many conditions [54,55,56,57]. Fibrinogen/fibrin deposition is associated with microglial activation and increased immune cell recruitment into the brain [54, 58]. In line with this finding, we showed microglial activation and increased fibrinogen deposition in the brains of CKD mice (Fig. 3). Activated microglia modulate expression of tight junction proteins essential for BBB integrity [59], which may further exacerbate BBB disruption and CMH formation.
Our assessment of microvascular integrity in vitro has relied on TEER and tracer permeability measurements. TEER is the measurement of electrical resistance across a cellular monolayer to evaluate integrity of the endothelial monolayer. In our in vitro study with ihBMEC, TEER was elevated in the first 24 h due to the trophic factors present in the serum and reached the highest value at 48 h. Incubation with CKD serum caused injury to the monolayer, as shown by a reduction in TEER after serum treatment initiation (Fig. 5A). This is consistent with our previous findings from an in vitro study incubating mouse brain endothelial cells (bEnd.3) with CKD serum [13]. Furthermore, the decrease in TEER was accompanied by a two-fold increase in sodium fluorescein permeability across the ihBMEC monolayer (Fig. 5B). Sodium fluorescein is a small molecular weight tracer (MW: 376 Da) that more readily diffuses through the BBB than larger molecular weight tracers, and therefore has served as a common marker for altered permeability [26]. Previously, we showed an inverse correlation between sodium fluorescein permeability and TEER measurements across the ihBMEC monolayer [26]. Together, impairment of the monolayer induced by CKD-derived serum factors allows for increased passage of sodium fluorescein across the monolayer, suggesting disruption of the endothelial monolayer and consistent with BBB injury observed with fibrinogen immunostaining.
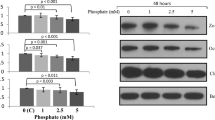
The murine gut microbiome changes with aging and is associated with alterations in microbial carbohydrate metabolism, decreased fecal short-chain fatty acids and decreased cobalamin and biotin biosynthesis [60, 61]. In the current study, we examined well-known gut-derived uremic toxins that are generated in the CKD milieu. These toxins are derived from amino acid catabolism by intestinal microbiota and are associated with systemic inflammation and vascular injury in CKD [62,63,64,65]. The exact uremic toxins that contribute to BBB disruption and whether the effects are results of an individual toxin or a combination of several toxins remain largely unknown. We have previously shown CKD serum from dialysis patients caused marked drop in TEER, and urea was one of the key uremic toxins. Exposure of bEnd.3 cells to urea at concentrations approximate to the values measured in dialysis patients reduced TEER in a dose-dependent manner [13]. In the current in vitro study, this was further investigated by exposing bEnd.3 mouse brain endothelial cells with various gut-derived uremic toxins alone and in combination, with TEER measurements every 24 h. We demonstrated that uremic toxins (i.e., IS, PCS, and TMAO) in combination with urea and LPS exerted the most deleterious effects on the endothelial barrier. TEER was significantly lower in the 3Toxins + Urea + LPS group compared with the control group, and eventually declined to a level close to the baseline values at day 4 (Fig. 5D). These findings suggest gut-derived uremic toxins aggravate urea/LPS-induced bEnd.3 endothelial barrier dysfunction, again consistent with findings of BBB injury observed with fibrinogen immunostaining.
Despite these multiple findings, this study has some limitations. The mouse study suggests a key role of microglia in mediating CMH formation in CKD animals. It should be noted that the use of CSF1R inhibitor PLX3397 eliminates microglia in the parenchyma, but also affects the number of non-parenchymal macrophages in the perivascular spaces, the choroid plexus, and the meninges [66], which are known to cause neurovascular dysfunction [67]. Our findings do not rule-out a contribution of perivascular macrophages to CMH formation [68], a subject that warrants further investigation. The binary morphological categorization of microglia into resting and activated states may be an oversimplification. Microglia intermediate between the two states may adopt various functions in immune cell recruitment and activation, cell proliferation, and phagocytosis as their morphology changes [69]. Note that the vascular source of CMH in uremic milieu remains unclear, as iron uptake into the brain is highly regulated by BBB. Mouse models and human postmortem studies of aging, hypertension, and Alzheimer’s disease have suggested a capillary source of CMH involving BBB disruption [14, 70,71,72], consistent with the findings from the in vitro CKD models we investigated. However, these findings should be interpreted with caution, because CMH may develop via a mechanism independent of capillary injury [73,74,75]. To expand our knowledge on the vascular source of CMH, we have developed a semi-automated approach to characterize microvascular network in three-dimensional (3D) imaging of mouse brains [76]; this will enable us to visualize the co-localization of fluorescently labeled microvascular network and Prussian blue-positive CMH and therefore, identify the vascular origin of CMH. In addition, the issue of cognitive decline with CKD is important, as we have emphasized [77]. However, the experiments in our manuscript were neither designed nor powered to address this issue. In terms of in vitro BBB models, recent studies show that ihBMEC have a mixed endothelial–epithelial transcriptional profile [26, 78, 79], and we therefore used two different brain endothelial cell culture systems, i.e., ihBMEC and bEnd.3 cells, for a better understanding of the mechanistic role of microvascular endothelial function in CKD-induced CMH formation.
Conclusions
Adenine-induced CKD promotes the development of CMH in aged C57BL/6J mice independent of blood pressure, likely via microglial activation and BBB disruption. Extent of CMH development is directly proportional to degree of renal insufficiency. Moreover, serum-derived factors in CKD disrupt endothelial monolayer by reducing TEER and enhancing the passage of sodium fluorescein across the monolayer. Gut-derived uremic toxins (i.e., IS, PCS, and TMAO) aggravated urea/LPS-induced endothelial barrier dysfunction by producing a marked drop in TEER, supporting the key role of uremic toxins in CKD-specific mechanisms that contribute to microvascular dysfunction. These findings indicate that CKD provokes microvascular injury leading to CMH formation in this model and suggest that CKD makes an important contribution to cerebral small vessel disease.
Availability of data and materials
Data are available upon reasonable request.
Abbreviations
- ABC:
-
Avidin–biotin-peroxidase
- BBB:
-
Blood–brain barrier
- bFGF:
-
Basic fibroblast growth factor
- CCK-8:
-
Cell Counting Kit-8
- CKD:
-
Chronic kidney disease
- CMH:
-
Cerebral microhemorrhages
- CSF1R:
-
Colony-stimulating factor 1 receptor
- DAB:
-
3,3′-Diaminobenzidine
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- DMEM/Ham’s F12:
-
Dulbecco's Modified Eagle Medium/Ham’s nutrient mixture F-12
- FBS:
-
Fetal bovine serum
- GFAP:
-
Glial fibrillary acidic protein
- hESFM:
-
Human endothelial serum-free medium
- ihBMEC:
-
Human brain microvascular endothelial cells
- IMR90-4:
-
IMR90 clone 4 line
- iPSC:
-
Human induced pluripotent stem cell
- IS:
-
Indoxyl sulfate
- LC–MS/MS:
-
Liquid chromatography with tandem mass spectrometry
- LDL:
-
Low-density lipoprotein
- LPS:
-
Lipopolysaccharide
- MEM–NEAA:
-
Minimum essential medium–nonessential amino acids
- PBS:
-
Phosphate-buffered saline
- PCS:
-
P-cresyl sulfate
- ROCK:
-
Rho-associated protein kinase
- RT:
-
Room temperature
- TEER:
-
Transendothelial electrical resistance
- TMAO:
-
Trimethylamine-N-oxide
- vWF:
-
Von Willebrand factor
References
Centers for disease control and prevention. Chronic kidney disease in the United States 2021. Atlanta: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2021.
Etgen T, Chonchol M, Förstl H, Sander D. Chronic kidney disease and cognitive impairment: a systematic review and meta-analysis. Am J Nephrol. 2012;35:474–82.
Masson P, Kelly PJ, Craig JC, Lindley RI, Webster AC. Risk of stroke in patients with ESRD. Clin J Am Soc Nephrol. 2015;10:1585–92.
Vanent KN, Leasure AC, Acosta JN, Kuohn LR, Woo D, Murthy SB, Kamel H, Messé SR, Mullen MT, Cohen JB, et al. Association of chronic kidney disease with risk of intracerebral hemorrhage. JAMA Neurol. 2022;79:911–8.
Chiu YL, Tsai HH, Lai YJ, Tseng HY, Wu YW, Peng YS, Chiu CM, Chuang YF. Cognitive impairment in patients with end-stage renal disease: accelerated brain aging? J Formos Med Assoc. 2019;118:867–75.
Donkor ES. Stroke in the 21(st) century: a snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat. 2018;2018:3238165.
Vinters HV, Magaki SD, Williams CK. Neuropathologic findings in chronic kidney disease (CKD). J Stroke Cerebrovasc Dis. 2021;30: 105657.
Lau WL, Huisa BN, Fisher M. The cerebrovascular-chronic kidney disease connection: perspectives and mechanisms. Transl Stroke Res. 2017;8:67–76.
Poels MM, Vernooij MW, Ikram MA, Hofman A, Krestin GP, van der Lugt A, Breteler MM. Prevalence and risk factors of cerebral microbleeds: an update of the Rotterdam scan study. Stroke. 2010;41:S103-106.
Chai C, Wang Z, Fan L, Zhang M, Chu Z, Zuo C, Liu L, Mark Haacke E, Guo W, Shen W, **a S. Increased number and distribution of cerebral microbleeds is a risk factor for cognitive dysfunction in hemodialysis patients: a longitudinal study. Medicine. 2016;95: e2974.
Ovbiagele B, Wing JJ, Menon RS, Burgess RE, Gibbons MC, Sobotka I, German L, Shara NM, Fernandez S, Jayam-Trouth A, et al. Association of chronic kidney disease with cerebral microbleeds in patients with primary intracerebral hemorrhage. Stroke. 2013;44:2409–13.
Shima H, Mori T, Ooi M, Sonoda M, Shoji T, Ishimura E, Okamura M, Ishizaka N, Inaba M. Silent cerebral microbleeds and longitudinal risk of renal and cardiovascular events in patients with CKD. Clin J Am Soc Nephrol. 2016;11:1557–65.
Lau WL, Nunes ACF, Vasilevko V, Floriolli D, Lertpanit L, Savoj J, Bangash M, Yao Z, Shah K, Naqvi S, et al. Chronic kidney disease increases cerebral microbleeds in mouse and man. Transl Stroke Res. 2020;11:122–34.
Sumbria RK, Grigoryan MM, Vasilevko V, Paganini-Hill A, Kilday K, Kim R, Cribbs DH, Fisher MJ. Aging exacerbates development of cerebral microbleeds in a mouse model. J Neuroinflammation. 2018;15:69.
Figuer A, Bodega G, Tato P, Valera G, Serroukh N, Ceprian N, de Sequera P, Morales E, Carracedo J, Ramírez R, Alique M. Premature aging in chronic kidney disease: the outcome of persistent inflammation beyond the bounds. Int J Environ Res Public Health. 2021;18:8044.
Xu KY, **a GH, Lu JQ, Chen MX, Zhen X, Wang S, You C, Nie J, Zhou HW, Yin J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci Rep. 2017;7:1445.
McIntyre CW, Harrison LE, Eldehni MT, Jefferies HJ, Szeto CC, John SG, Sigrist MK, Burton JO, Hothi D, Korsheed S, et al. Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:133–41.
Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, Argiles A. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 2012;23:1258–70.
Assem M, Lando M, Grissi M, Kamel S, Massy ZA, Chillon JM, Hénaut L. The impact of uremic toxins on cerebrovascular and cognitive disorders. Toxins. 2018;10:303.
Fox JG, Barthold S, Davisson M, Newcomer CE, Quimby FW, Smith A. The mouse in biomedical research: normative biology, husbandry, and models. Amsterdam: Elsevier; 2006.
Passos GF, Kilday K, Gillen DL, Cribbs DH, Vasilevko V. Experimental hypertension increases spontaneous intracerebral hemorrhages in a mouse model of cerebral amyloidosis. J Cereb Blood Flow Metab. 2016;36:399–404.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–97.
Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, **e Z, Malhotra D, Kolodkin NI, et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–95.
Lau WL, Khazaeli M, Savoj J, Manekia K, Bangash M, Thakurta RG, Dang A, Vaziri ND, Singh B. Dietary tetrahydrocurcumin reduces renal fibrosis and cardiac hypertrophy in 5/6 nephrectomized rats. Pharmacol Res Perspect. 2018;6: e00385.
Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology. 2016;233:1543–57.
Sun J, Ou W, Han D, Paganini-Hill A, Fisher MJ, Sumbria RK. Comparative studies between the murine immortalized brain endothelial cell line (bEnd.3) and induced pluripotent stem cell-derived human brain endothelial cells for paracellular transport. PLoS ONE. 2020;17: e0268860.
Rodrigues WF, Miguel CB, Napimoga MH, Oliveira CJ, Lazo-Chica JE. Establishing standards for studying renal function in mice through measurements of body size-adjusted creatinine and urea levels. Biomed Res Int. 2014;2014: 872827.
Keane L, Antignano I, Riechers SP, Zollinger R, Dumas AA, Offermann N, Bernis ME, Russ J, Graelmann F, McCormick PN, et al. mTOR-dependent translation amplifies microglia priming in aging mice. J Clin Invest. 2021. https://doi.org/10.1172/JCI155208.
Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. J Am Soc Nephrol. 2000;11:319–29.
Hecking M, Bieber BA, Ethier J, Kautzky-Willer A, Sunder-Plassmann G, Säemann MD, Ramirez SP, Gillespie BW, Pisoni RL, Robinson BM, Port FK. Sex-specific differences in hemodialysis prevalence and practices and the male-to-female mortality rate: the dialysis outcomes and practice patterns study (DOPPS). PLoS Med. 2014;11: e1001750.
Ricardo AC, Yang W, Sha D, Appel LJ, Chen J, Krousel-Wood M, Manoharan A, Steigerwalt S, Wright J, Rahman M, et al. Sex-related disparities in CKD progression. J Am Soc Nephrol. 2019;30:137–46.
Minutolo R, Gabbai FB, Chiodini P, Provenzano M, Borrelli S, Garofalo C, Bellizzi V, Russo D, Conte G, De Nicola L. Sex differences in the progression of CKD among older patients: pooled analysis of 4 cohort studies. Am J Kidney Dis. 2020;75:30–8.
Antlanger M, Noordzij M, van de Luijtgaarden M, Carrero JJ, Palsson R, Finne P, Hemmelder MH, Aresté-Fosalba N, Reisæter AV, Cases A, et al. Sex differences in kidney replacement therapy initiation and maintenance. Clin J Am Soc Nephrol. 2019;14:1616–25.
Hödlmoser S, Winkelmayer WC, Zee J, Pecoits-Filho R, Pisoni RL, Port FK, Robinson BM, Ristl R, Krenn S, Kurnikowski A, et al. Sex differences in chronic kidney disease awareness among US adults, 1999 to 2018. PLoS ONE. 2020;15: e0243431.
Valdivielso JM, Jacobs-Cachá C, Soler MJ. Sex hormones and their influence on chronic kidney disease. Curr Opin Nephrol Hypertens. 2019;28:1–9.
Verzola D, Gandolfo MT, Salvatore F, Villaggio B, Gianiorio F, Traverso P, Deferrari G, Garibotto G. Testosterone promotes apoptotic damage in human renal tubular cells. Kidney Int. 2004;65:1252–61.
Usselman CW, Stachenfeld NS, Bender JR. The molecular actions of oestrogen in the regulation of vascular health. Exp Physiol. 2016;101:356–61.
Frick KM, Burlingame LA, Arters JA, Berger-Sweeney J. Reference memory, anxiety and estrous cyclicity in C57BL/6NIA mice are affected by age and sex. Neuroscience. 2000;95:293–307.
Mazumder MK, Paul R, Bhattacharya P, Borah A. Neurological sequel of chronic kidney disease: from diminished Acetylcholinesterase activity to mitochondrial dysfunctions, oxidative stress and inflammation in mice brain. Sci Rep. 2019;9:3097.
Hirotsu C, Tufik S, Ribeiro DA, Alvarenga TA, Andersen ML. Genomic damage in the progression of chronic kidney disease in rats. Brain Behav Immun. 2011;25:416–22.
** WS, Shen LL, Bu XL, Zhang WW, Chen SH, Huang ZL, **ong JX, Gao CY, Dong Z, He YN, et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017;134:207–20.
Mayne K, White JA, McMurran CE, Rivera FJ, de la Fuente AG. Aging and neurodegenerative disease: is the adaptive immune system a friend or foe? Front Aging Neurosci. 2020;12: 572090.
Xue Y, Nie D, Wang LJ, Qiu HC, Ma L, Dong MX, Tu WJ, Zhao J. Microglial polarization: novel therapeutic strategy against ischemic stroke. Aging Dis. 2021;12:466–79.
Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflammation. 2022;19:206.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42:318–33.
Angelova DM, Brown DR. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J Neurochem. 2019;151:676–88.
Hénaut L, Grissi M, Brazier F, Assem M, Poirot-Leclercq S, Lenglet G, Boudot C, Avondo C, Boullier A, Choukroun G, et al. Cellular and molecular mechanisms associated with ischemic stroke severity in female mice with chronic kidney disease. Sci Rep. 2019;9:6432.
Li LC, Chen WY, Chen JB, Lee WC, Chang CC, Tzeng HT, Huang CC, Chang YJ, Yang JL. The AST-120 recovers uremic toxin-induced cognitive deficit via NLRP3 inflammasome pathway in astrocytes and microglia. Biomedicines. 2021;9:1252.
Savage JC, Carrier M, Tremblay M-È. Morphology of microglia across contexts of health and disease. In: Verkhratsky A, Garaschuk O, editors. Microglia: methods and protocols. New York: Springer; 2019.
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25.
Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, Nelson D, Takechi R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 2015;12:2.
Zhao Y, Liu Y, Xu Y, Li K, Zhou L, Qiao H, Xu Q, Zhao J. The role of ferroptosis in blood-brain barrier injury. Cell Mol Neurobiol. 2022. https://doi.org/10.1007/s10571-022-01197-5.
Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227.
Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204:1999–2008.
Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66:695–709.
Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. 2010;30:5843–54.
Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, Vagena E, Bedard C, Machado MR, Rios Coronado PE, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun. 2015;6:8164.
Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS ONE. 2014;9: e101815.
Langille MG, Meehan CJ, Koenig JE, Dhanani AS, Rose RA, Howlett SE, Beiko RG. Microbial shifts in the aging mouse gut. Microbiome. 2014;2:50.
You X, Dadwal UC, Lenburg ME, Kacena MA, Charles JF. Murine gut microbiome meta-analysis reveals alterations in carbohydrate metabolism in response to aging. GmSystems. 2022;7: e0124821.
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, Desantis TZ, Ni Z, Nguyen TH, Andersen GL. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83:308–15.
Mishima E, Fukuda S, Mukawa C, Yuri A, Kanemitsu Y, Matsumoto Y, Akiyama Y, Fukuda NN, Tsukamoto H, Asaji K, et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017;92:634–45.
Lau WL, Savoj J, Nakata MB, Vaziri ND. Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci (Lond). 2018;132:509–22.
Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. 2018;14:442–56.
Mildenberger W, Stifter SA, Greter M. Diversity and function of brain-associated macrophages. Curr Opin Immunol. 2022;76: 102181.
Santisteban MM, Ahn SJ, Lane D, Faraco G, Garcia-Bonilla L, Racchumi G, Poon C, Schaeffer S, Segarra SG, Körbelin J, et al. Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension. Hypertension. 2020;76:795–807.
Polfliet MM, Goede PH, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol. 2001;116:188–95.
Karperien A, Ahammer H, Jelinek HF. Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci. 2013;7:3.
Fisher M, French S, Ji P, Kim RC. Cerebral microbleeds in the elderly: a pathological analysis. Stroke. 2010;41:2782–5.
Craggs LJ, Yamamoto Y, Deramecourt V, Kalaria RN. Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol. 2014;24:495–509.
Kalaria RN. Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer’s disease. Ann NY Acad Sci. 1997;826:263–71.
Chang R, Castillo J, Zambon AC, Krasieva TB, Fisher MJ, Sumbria RK. Brain endothelial erythrophagocytosis and hemoglobin transmigration across brain endothelium: implications for pathogenesis of cerebral microbleeds. Front Cell Neurosci. 2018;12:279.
Wadi LC, Grigoryan MM, Kim RC, Fang C, Kim J, Corrada MM, Paganini-Hill A, Fisher MJ. Mechanisms of cerebral microbleeds. J Neuropathol Exp Neurol. 2020;79:1093–9.
Shih AY, Hyacinth HI, Hartmann DA, van Veluw SJ. Rodent models of cerebral microinfarct and microhemorrhage. Stroke. 2018;49:803–10.
**e DF, Crouzet C, Lopresti K, Wang Y, Robinson C, Jones W, Muqolli F, Pai A, Fang C, Cribbs DH. Semi-automated protocol to quantify and characterize fluorescent, 3D vascular images. bioRxiv. 2022. https://doi.org/10.1101/2022.05.05.490827.
Lau WL, Fisher M. New insights into cognitive decline in chronic kidney disease. Nat Rev Nephrol. 2022. https://doi.org/10.1038/s41581-022-00656-y.
Delsing L, Dönnes P, Sánchez J, Clausen M, Voulgaris D, Falk A, Herland A, Brolén G, Zetterberg H, Hicks R, Synnergren J. Barrier properties and transcriptome expression in human iPSC-derived models of the blood-brain barrier. Stem Cells. 2018;36:1816–27.
Lu TM, Houghton S, Magdeldin T, Durán JGB, Minotti AP, Snead A, Sproul A, Nguyen D-HT, **ang J, Fine HA, et al. Pluripotent stem cell-derived epithelium misidentified as brain microvascular endothelium requires ETS factors to acquire vascular fate. Proc Natl Acad Sci. 2021;118: e2016950118.
Acknowledgements
Not applicable.
Funding
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke under award numbers R01NS20989 (MJF and DHC), R01NS113337 (WLL), and by the National Institute of Aging under award numbers R01AG072896 and R01AG062840 (RKS) of the National Institutes of Health. Approximately $400K (100%) of Federal funds supported this project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
CF, WLL, JS, RC, APH, RKS, DHC, and MF designed the study, analyzed the results, and wrote and revised the manuscript. CF, JS, RC, JL, and HL performed the experiments and collected data. CF, JS, RC, AV, DL, YHH and YZ participated in the data acquisition, analysis, and interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures described in this article were approved the Institutional Animal Care and Use Committee or Institutional Review Board at the University of California, Irvine.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fang, C., Lau, W.L., Sun, J. et al. Chronic kidney disease promotes cerebral microhemorrhage formation. J Neuroinflammation 20, 51 (2023). https://doi.org/10.1186/s12974-023-02703-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02703-2