
Abstract
Background
Chemotherapy-induced peripheral neuropathy (CIPN) is a serious side effect of chemotherapy with poorly understood mechanisms and few treatments. High-mobility group box 1 (HMGB1)-induced neuroinflammation is the main cause of CIPN. Here, we aimed to illustrate the role of the macrophage scavenger receptor A1 (SR-A1) in HMGB1 clearance and CIPN resolution.
Methods
Oxaliplatin (L-OHP) was used to establish a CIPN model. Recombinant HMGB1 (rHMGB1) (his tag) was used to evaluate the phagocytosis of HMGB1 by macrophages.
Results
In the clinic, HMGB1 expression and MMP-9 activity were increased in the plasma of patients with CIPN. Plasma HMGB1 expression was positively correlated with the cumulative dose of L-OHP and the visual analog scale. In vitro, engulfment and degradation of rHMGB1 increased and inflammatory factor expression decreased after AMP-activated protein kinase (AMPK) activation. Neutralizing antibodies, inhibitors, or knockout of SR-A1 abolished the effects of AMPK activation on rHMGB1 engulfment. In vivo, AMPK activation increased SR-A1 expression in the dorsal root ganglion, decreased plasma HMGB1 expression and MMP-9 activity, and attenuated CIPN, which was abolished by AMPK inhibition or SR-A1 knockout in the CIPN mice model.
Conclusion
Activation of the AMPK/SR-A1 axis alleviated CIPN by increasing macrophage-mediated HMGB1 engulfment and degradation. Therefore, promoting HMGB1 clearance may be a potential treatment strategy for CIPN.
Video abstract
Similar content being viewed by others

Background
Chemotherapy-induced peripheral neuropathy (CIPN) is a major dose-limiting side effect of chemotherapeutic agents, including platinum drugs, taxanes, epothilones, and vinca alkaloids [1]. Up to 76.5% of patients develop CIPN during and after treatment with chemotherapeutic drugs [2], especially oxaliplatin (up to 89%) [3]. The manifestation is peripheral neuropathy with a ‘stocking and glove’ distribution, characterized by sensory loss, paresthesia, dysesthesia, numbness, and tingling, often aggravated by neuropathic pain [4]. Unfortunately, there is no effective treatment to date [5]. Thus, there is an urgent need to explore the underlying mechanisms and develop effective treatment strategies.
Neuroinflammation is widely considered the major mechanism of CIPN [6, 7]. Excessive expression of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, etc.) causes peripheral sensitization and subsequent central sensitization, leading to CIPN [8, 9]. High mobility group box 1 (HMGB1), a damage-associated molecular pattern (DAMP), can induce the expression of pro-inflammatory cytokines and act as an initiator and amplifier of neuroinflammation in neuropathic pain [10, 11]. Our previous study demonstrated that oxaliplatin induced the release of HMGB1 from neurons and Raw 264.7 cells, which activates MMP-9 and induces and aggravates CIPN [12], suggesting that HMGB1 could be a potential target for CIPN therapy.
Endocytosis of ligand molecules is considered a mechanism of signal attenuation via receptor and ligand clearance from the cell surface [13]. Macrophage scavenger receptor A1 (SR-A1), a member of the scavenger receptor family, plays multiple roles in the phagocytosis of macrophages [Cell staining BMDMs were plated in glass bottom cell culture dishes and activated with LPS (E. coli 0111:B4; 100 ng/ml; Sigma) + IFN-γ (50 ng/ml; Peprotech) for 24 h. The cells were then treated with or without AICAR (300 μM) for 15 min before being co-cultured with Human HMGB1 protein (his tag) (10 nM). BMDMs were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. After blocking with 10% donkey serum in PBS for 2 h, the coverslips with BMDM cells were incubated at 4℃ with the His Tag antibody (1:200) diluted in PBS overnight. The coverslips were then exposed to fluorescent secondary antibody (1:300, at room temperature for 2 h) and rinsed three times with PBS. 4′,6-Diamidino-2-phenylindole (DAPI) is a fluorescent DNA dye that marks the nucleus. Confocal microscopy was performed using a Carl Zeiss LSM710 confocal system. Under deep anesthesia by an intraperitoneal injection of sodium pentobarbital (40 mg/kg), the animals were perfused transcardially with normal saline followed by 4% paraformaldehyde in 0.1 M PB, pH7.4, each for 20 min. Then, L4 and L5 lumbar segments were dissected and post-fixed in 4% paraformaldehyde. The embedded blocks were sectioned at a thickness of 25 μm. Sections from each group (five animals in each group) were incubated with rabbit antibodies for c-Fos (1:200) and CGRP (1:200). The free-floating sections were washed with PBS and incubated with secondary antibody (1:300; Jackson Laboratories, USA) for 2 h at room temperature. After washing three times with PBS, the samples were examined under a confocal microscope (Olympus FV1000 confocal system, Olympus, Japan) for morphological details of immunofluorescence staining. The examinations were performed blindly. Images were randomly coded, and fluorescence intensities were analyzed using ImageJ software. The average green fluorescence intensity of each pixel was normalized to the background intensity of the same image. Total RNA was extracted from BMDMs using TRIzol reagent (Invitrogen, CA, USA). Isolated RNA was reverse transcribed into cDNA using HiScript II Q RT SuperMix for qPCR (Vazyme Biotech Co., Ltd.) following the standard protocol. Quantitative real-time PCR (qPCR) was performed using ChamQ SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) with a QuantStudio 5 Real-Time PCR Detection System (Thermo Fisher Scientific). The relative expression levels of TNF-α, IL-1β, and IL-6 were calculated and quantified using the 2 − ΔΔCt method after normalization with a reference. All the primers used are listed in Additional file 1: Table S2. The procedure was according to the instruction. In short, DRG samples were collected on the 7 th day after the first administration of L-OHP (3 mg/kg, i.p.) or vehicle (n = 4). Nitric acid (analytical purity) was added into the DRG samples to completely dissolve the samples (37 ℃ water bath for 10 min). Hydrogen peroxide was added into the solution (room temperature for 10 min). Inductively coupled plasma emission mass spectrometer (ICP-MS 7800) was started to detect platinum content in samples and collect and analyze data. GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA) was used to conduct all the statistical analyses. Alteration of expression of the proteins and the behavioral responses were tested with one-way analysis of variance (ANOVA), and the differences in latency over time among groups were tested with two-way ANOVA, followed by Bonferroni’s post hoc tests. Unpaired Student’s t-test was used to analyze differences between the two groups. The correlation between visual analog scale (VAS) and L-OHP dose, plasma HMGB1 accumulation and L-OHP dose, and VAS and plasma HMGB1 accumulation in CIPN patients were analyzed using linear regression analysis. Results are expressed as mean ± SEM. p values < 0.05 were considered significant.Immunofluorescence assay
Quantitative PCR (qPCR)
Determination of platinum concentration in DRG
Statistics
Results
L-OHP-induced CIPN is associated with a high level of HMGB1 accumulation and MMP-9/2 activity
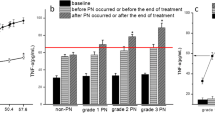
To explore the critical role of HMGB1 in the development of CIPN, we evaluated the concentration of HMGB1 and the activity of MMP-9 in the plasma of patients with CIPN and mice. MMP-9 is a downstream target enzyme of HMGB1 and is an important marker of pain. As shown in Fig. 1A and B, HMGB1 concentration and MMP-9 activity in the plasma of patients significantly increased after L-OHP treatment. There is a positive correlation between visual analogue scale (VAS) and L-OHP dose (r = 0.8594, p < 0.001, data not show), plasma HMGB1 accumulation and L-OHP dose, VAS and plasma HMGB1 accumulation in CIPN patients respectively (Fig. 1C and D). Additionally, mice were treated with L-OHP (3 mg/kg, i.p., five consecutive days at a total dose of 15 mg/kg) to establish CIPN mice models(Fig. 1E). L-OHP significantly decreased the mechanical thresholds of the mice on day 2, and the induced allodynia lasted until day 14 (Fig. 1F). Compared to the vehicle group, HMGB1 accumulation and MMP-9 activity were dramatically increased in CIPN mouse plasma (Fig. 1G and H). These data suggest that excessive accumulation of HMGB1 plays a vital role in CIPN development.

HMGB1 accumulation and MMP-9/2 activity are increased in the plasma of CIPN patients and mice. A, B Plasma HMGB1 content (n = 20) and MMP-9 (n = 12) activity in cancer patients before L-OHP treatment (Control) and after the last L-OHP treatment (CIPN). The correlation between plasma HMGB1 and L-OHP dose (C) and plasma HMGB1 and VAS (D), respectively. VAS was performed after the last L-OHP treatment (n = 20). E The general scheme of experiments in vivo. F Mechanical thresholds of mice received L-OHP administration (3 mg/kg, i.p.) or vehicle for five consecutive days (n = 6). G and H HMGB1 content and MMP-9/2 activities in plasma of mice measured by western blotting and Gelatin zymography (n = 3). Plasma was collected from mice on the 14th day after the first administration of L-OHP (3 mg/kg, i.p.) or vehicle. Representative bands and a data summary (n = 3 for each group) are shown. A significant difference was revealed following unpaired Student’s t-test (A, B, G and H) or two-way ANOVA (F) or Linear regression analysis (C and D) (*p < 0.05, **p < 0.01 and ***p < 0.001 vs. Control; Bonferroni post hoc tests)
AMPK activation increases macrophages’ engulfment of HMGB1 and reduces inflammation
To find a solution to clear HMGB1, macrophage phagocytosis has attracted our attention for the endocytosis of HMGB1. Recombinant HMGB1 (rHMGB1) (his tag) was used to evaluate macrophage phagocytosis. As shown in Fig. 2A, pretreatment with L-OHP (0.5, 1, or 2 h) decreased the amount of rHMGB1 (his tag) in mouse BMDMs after being co-cultured with rHMGB1 (his tag) for 60 min. Considering the regulatory effect of AMPK on macrophage phagocytosis [21, 12, 29]. Another study validates our conclusion in clinic and showes that NAC could reduce the incidence of the neuropathy induced by oxaliplatin and delay its occurrence in patients with gastric or colorectal cancers, although the mechanism has not been clarified [30]. In this study, for the first time, we found that in the plasma of patients and mice, L-OHP-induced mechanical allodynia was associated with increased HMGB1 accumulation and MMP-9/2 activity (Fig. 1A and B). Additionally, we found a positive correlation between plasma HMGB1 accumulation and VAS and plasma HMGB1 accumulation and L-OHP cumulative dose in patients with CIPN (Fig. 1C and D). Considering the important role of HMGB1, we further investigated the pathogenesis of CIPN from another perspective: scavenging HMGB1.
Is there an endogenous mechanism underlying HMGB1 clearance? Macrophages are known to act as ‘scavengers,’ clearing pathogens, bacteria, apoptotic cell debris, etc. [31]. Macrophages also play critical roles in HMGB1-mediated inflammatory response [32]. Evidence has confirmed that HMGB1 triggers dynamin-dependent endocytosis in macrophages [33]. The results showed that macrophages could endocytose HMGB1, which was significantly reduced by L-OHP treatment (Fig. 2A). Interestingly, the inhibition of HMGB1 engulfment was associated with a reduction in p-AMPK in the L-OHP group (Fig. 2B and C). However, the role of AMPK in the macrophage endocytosis of HMGB1 remains unclear. Since our previous study demonstrated that AMPK activation attenuates neuropathic pain in rats [19], we speculated that AMPK activation may attenuate L-OHP-induced peripheral neuropathy.
We investigated the relationship between AMPK and macrophage phagocytosis by HMGB1 cells. The data showed that AMPK activation significantly increased the engulfment of HMGB1 not only in mouse BMDMs but also in human PBMC-derived primary macrophages (Fig. 2D–I). We also found that AMPK activation decreased the mRNA levels of inflammatory cytokines induced by HMGB1 (Fig. 2J), suggesting that AMPK activation may decrease HMGB1 accumulation and inflammation. Additionally, two other AMPK activators were used for the experiments with mouse BMDMs (Fig. 3A). However, the AMPK activator, metformin, did not increase the amount of rHMGB1 (his tag) in BMDMs. Two possible mechanisms exist: (1) metformin does not promote rHMGB1 (his tag) internalization, and (2) metformin promotes rHMGB1 (his tag) internalization and degradation. The results showed that lysosomal inhibitors, but not protease inhibitors, could significantly inhibit the degradation of rHMGB1 (his tag) in BMDMs treated with metformin, AICAR, and A769662 (Fig. 3B and C). Combined with the lysotracker data (Fig. 3D), the results indicate that AMPK activation could increase the engulfment and degradation of HMGB1 in BMDMs lysosomes.
Therefore, it is necessary to explore the mechanism of HMGB1 engulfment in relation to AMPK. Macrophage endocytosis includes (TLRs, RAGE, SR, etc.)-mediated endocytosis [23, 34, 35]. Studies have shown that DAMPs, especially peroxiredoxin clearance in the brain by infiltrating mononuclear phagocytes, are beneficial for the resolution of sterile inflammation and treatment of stroke, where SR-A1 and HMGB1 are mentioned [16]. However, the study did not provide direct evidence that SR-A1 mediates the internalization of HMGB1 in vitro but showed that HMGB1 content in the brain was regulated by SR-A1 in vivo, suggesting the possibility that SR-A1 is involved in macrophage phagocytosis of HMGB1. In this study, we activated AMPK using agonists and performed further studies. We found that SR-A1 inhibitor, neutralizing antibody, and SR-A1 knockout significantly inhibited the engulfment of HMGB1 induced by AICAR or metformin (Fig. 4). Additionally, AMPK activation could promote SR-A1 translocation to the cytomembrane, the process by which PKC participates (Fig. 5).
It has been reported that HMGB1 endocytosis by macrophages could lead to lysosomal destabilization and macrophage pyroptosis via the RAGE-mediated signaling pathway [33]. In this study, we showed that the engulfment of HMGB1 promoted by AMPK activation was abolished by the lysosomal inhibitor CQ but not by the RAGE inhibitor FPS-ZM1 (Figs. 3C and 4B). Lysosomes maintained their morphology and co-localized with HMGB1 1 h after rHMGB1 (his tag) administration (Fig. 3D), suggesting that the AMPK/SR-A1 axis-mediated engulfment of HMGB1 may not induce macrophage pyroptosis.
We further verified the analgesic effects of AMPK activation on CIPN in vivo. The AMPK activator, metformin, attenuated L-OHP-induced mechanical allodynia, decreased HMGB1 accumulation and MMP-9 activity in CIPN mouse blood, and reduced the expression of CGRP (an indicator of pain) and c-fos (a marker of nociceptive neuron activation) in the spinal cord of CIPN mice (Fig. 6). In addition, considering that metformin is a substrate of OCT2 transporter as well as oxaliplatin, we measured the drug-drug interaction (competition for OCT2 transport) happened between metformin and oxaliplatin. The data was shown in Additional file 1: Fig. S1, metformin did not affect the level of L-OHP in the DRG of mice. We also investigated the mechanism underlying the analgesic effect of metformin. AMPK activation increased the expression of SR-A1 in the DRG of mice. AMPK inhibition, PKC inhibition, and SR-A1 knockout abolished the suppressive effects of metformin on mechanical allodynia, HMGB1 accumulation, and MMP-9 activity (Fig. 7) in CIPN mice in vivo.
Conclusion
In summary, we demonstrated that HMGB1 plays an important role in CIPN in animal experiments and clinics and provides metformin as a potential treatment for CIPN, which could activate the AMPK/p38/SR-A1 axis to promote HMGB1 clearance (Fig. 8).

Schematic model indicates that promoting SR-A1-mediated engulfment of HMGB1 by AMPK activation facilitates the resolution of inflammation and alleviates CIPN. L-OHP increases HMGB1 release from stress cells and inhibits macrophage endocytosis of HMGB1 via AMPK inactivation. HMGB1 accumulation subsequently induces MMP-9 release via the TLR4-mediated inflammatory signaling pathway and contributes to the progression of CIPN. AMPK agonists activate PKC and promote SR-A1 transfer to the membrane to mediate the engulfment of HMGB1 and degradation in lysosome, leading to the resolution of inflammation and attenuation of CIPN
Availability of data and materials
All data associated with this study are presented in the paper and/or Additional file 2 showed as Raw data.
Abbreviations
- CIPN:
-
Chemotherapy-induced peripheral neuropathy
- HMGB1:
-
High mobility group box 1
- VAS:
-
Visual analog scale
- AMPK:
-
AMP-activated protein kinase
- SR-A1:
-
Macrophage scavenger receptor A1
- L-OHP:
-
Oxaliplatin
- PKC:
-
Protein kinase C
- PBMC:
-
Peripheral blood mononuclear cells
- BMDMs:
-
Bone marrow-derived macrophages
- MMP:
-
Matrix metalloproteinase
References
Hertz DL, Childs DS, Park SB, Faithfull S, Ke Y, Ali NT, McGlown SM, Chan A, Grech LB, Loprinzi CL, et al. Patient-centric decision framework for treatment alterations in patients with chemotherapy-induced peripheral neuropathy (CIPN). Cancer Treat Rev. 2021;99: 102241.
Battaglini E, Goldstein D, Grimison P, McCullough S, Mendoza-Jones P, Park SB. Chemotherapy-induced peripheral neurotoxicity in cancer survivors: predictors of long-term patient outcomes. J Natl Compr Canc Netw. 2021;19:821–8.
Mizrahi D, Park SB, Li T, Timmins HC, Trinh T, Au K, Battaglini E, Wyld D, Henderson RD, Grimison P, et al. Hemoglobin, body mass index, and age as risk factors for paclitaxel- and oxaliplatin-induced peripheral neuropathy. JAMA Netw Open. 2021;4: e2036695.
Jordan B, Margulies A, Cardoso F, Cavaletti G, Haugnes HS, Jahn P, Le Rhun E, Preusser M, Scotte F, Taphoorn MJB, et al. Systemic anticancer therapy-induced peripheral and central neurotoxicity: ESMO-EONS-EANO clinical practice guidelines for diagnosis, prevention, treatment and follow-up. Ann Oncol. 2020;31:1306–19.
Loprinzi CL, Lacchetti C, Bleeker J, Cavaletti G, Chauhan C, Hertz DL, Kelley MR, Lavino A, Lustberg MB, Paice JA, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO guideline update. J Clin Oncol. 2020;38:3325–48.
McLeary F, Davis A, Rudrawar S, Perkins A, Anoopkumar-Dukie S. Mechanisms underlying select chemotherapeutic-agent-induced neuroinflammation and subsequent neurodegeneration. Eur J Pharmacol. 2019;842:49–56.
Lees JG, Makker PG, Tonkin RS, Abdulla M, Park SB, Goldstein D, Moalem-Taylor G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur J Cancer. 2017;73:22–9.
Hu LY, Mi WL, Wu GC, Wang YQ, Mao-Ying QL. Prevention and treatment for chemotherapy-induced peripheral neuropathy: therapies based on CIPN mechanisms. Curr Neuropharmacol. 2019;17:184–96.
Han Y, Smith MT. Pathobiology of cancer chemotherapy-induced peripheral neuropathy (CIPN). Front Pharmacol. 2013;4:156.
Das N, Dewan V, Grace PM, Gunn RJ, Tamura R, Tzarum N, Watkins LR, Wilson IA, Yin H. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 2016;17:1128–40.
Shibasaki M, Sasaki M, Miura M, Mizukoshi K, Ueno H, Hashimoto S, Tanaka Y, Amaya F. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain. 2010;149:514–21.
Gu H, Wang C, Li J, Yang Y, Sun W, Jiang C, Li Y, Ni M, Liu WT, Cheng Z, Hu L. High mobility group box-1-toll-like receptor 4-phosphatidylinositol 3-kinase/protein kinase B-mediated generation of matrix metalloproteinase-9 in the dorsal root ganglion promotes chemotherapy-induced peripheral neuropathy. Int J Cancer. 2020;146:2810–21.
Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol. 2011;23:393–403.
Bonilla DL, Bhattacharya A, Sha Y, Xu Y, **ang Q, Kan A, Jagannath C, Komatsu M, Eissa NT. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39:537–47.
Xu Z, Xu L, Li W, ** X, Song X, Chen X, Zhu J, Zhou S, Li Y, Zhang W, et al. Innate scavenger receptor-A regulates adaptive T helper cell responses to pathogen infection. Nat Commun. 2017;8:16035.
Shichita T, Ito M, Morita R, Komai K, Noguchi Y, Ooboshi H, Koshida R, Takahashi S, Kodama T, Yoshimura A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med. 2017;23:723–32.
Guo DY, Cao C, Zhang XY, **ang LX, Shao JZ. Scavenger receptor SCARA5 acts as an HMGB1 recognition molecule negatively involved in HMGB1-mediated inflammation in fish models. J Immunol. 2016;197:3198–213.
Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299–313.
Lu L, Pan C, Chen L, Hu L, Wang C, Han Y, Yang Y, Cheng Z, Liu WT. AMPK activation by peri-sciatic nerve administration of ozone attenuates CCI-induced neuropathic pain in rats. J Mol Cell Biol. 2017;9:132–43.
Bae HB, Zmijewski JW, Deshane JS, Tadie JM, Chaplin DD, Takashima S, Abraham E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011;25:4358–68.
Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-series reduce neutrophil extracellular traps. Blood. 2021;139(8):1222–33.
Wang S, Lin Y, **ong X, Wang L, Guo Y, Chen Y, Chen S, Wang G, Lin P, Chen H, et al. Low-dose metformin reprograms the tumor immune microenvironment in human esophageal cancer: results of a phase II clinical trial. Clin Cancer Res. 2020;26:4921–32.
Zhang W, Zhou Y, Li X, Xu X, Chen Y, Zhu R, Yin L. Macrophage-targeting and reactive oxygen species (ROS)-responsive nanopolyplexes mediate anti-inflammatory siRNA delivery against acute liver failure (ALF). Biomater Sci. 2018;6:1986–93.
Shen SQ, Lim G, You ZR, Ding WH, Huang PG, Ran CZ, Doheny J, Caravan P, Tate S, Hu K, et al. Gut microbiota is critical for the induction of chemotherapy-induced pain. Nat Neurosci. 2017;20:1213.
Illias AM, Gist AC, Zhang H, Kosturakis AK, Dougherty PM. Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain. 2018;159:1308–16.
Xu Y, Qian L, Zong G, Ma K, Zhu X, Zhang H, Li N, Yang Q, Bai H, Ben J, et al. Class A scavenger receptor promotes cerebral ischemic injury by pivoting microglia/macrophage polarization. Neuroscience. 2012;218:35–48.
Heishi M, Ichihara J, Teramoto R, Itakura Y, Hayashi K, Ishikawa H, Gomi H, Sakai J, Kanaoka M, Taiji M, Kimura T. Global gene expression analysis in liver of obese diabetic db/db mice treated with metformin. Diabetologia. 2006;49:1647–55.
Augusto PSA, Braga AV, Rodrigues FF, Morais MI, Dutra MMGB, Batista CRA, Melo ISF, Costa SOAM, Goulart FA, Coelho MM, Machado RR. Metformin antinociceptive effect in models of nociceptive and neuropathic pain is partially mediated by activation of opioidergic mechanisms. Eur J Pharmacol. 2019;858:172497.
Li J, Xu L, Deng X, Jiang C, Pan C, Chen L, Han Y, Dai W, Hu L, Zhang G, et al. N-acetyl-cysteine attenuates neuropathic pain by suppressing matrix metalloproteinases. Pain. 2016;157:1711–23.
Bondad N, Boostani R, Barri A, Elyasi S, Allahyari A. Protective effect of N-acetylcysteine on oxaliplatin-induced neurotoxicity in patients with colorectal and gastric cancers randomized, double blind, placebo-controlled, clinical trial. J Oncol Pharm Pract. 2020;26:1575–82.
Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–34.
Yang HA, Hreggvidsdottir HS, Palmblad K, Wang HC, Ochani M, Li JH, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–7.
Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, **ao G, Li S, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39.
Ghigo E, Kartenbeck J, Lien P, Pelkmans L, Capo C, Mege JL, Raoult D. Ameobal pathogen mimivirus infects macrophages through phagocytosis. PLoS Pathog. 2008;4:e1000087.
Prantner D, Nallar S, Vogel SN. The role of RAGE in host pathology and crosstalk between RAGE and TLR4 in innate immune signal transduction pathways. FASEB J. 2020;34:15659–74.
Acknowledgements
None.
Funding
This work was supported by the National Natural Science Foundation of China (82271252, 82204542, 81971047, and 81870870), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 21KJB310019, 21KJB310009), and Class A of key projects of Jiangsu Committee of Health (ZDA2020020).
Author information
Authors and Affiliations
Contributions
YX, FH, RMJ, and TTL performed western blotting and analyzed the results. YX, FH, TTL, CLP and XTZ performed the animal experiments, gelatin zymography, and H&E staining. YX, RMJ, TTL, YY, CJX, SRR, and CYJ carried out the cell cultures and immunofluorescence assay. YY, LH, QC, and WTL conceived the study, participated in its design and coordination, and helped to draft the manuscript. RM J, WF and YY help revised the manuscript. All the authors approved the final manuscript. YX, FH, RMJ, and WF contributed equally to the study. These four authors contributed the most to the article and ranked the authors according to their workloads. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures were performed in accordance with the regulations of the Ethics Committee of the International Association for the Study of Pain and Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China). All animal experiments were designed to minimize suffering and the number of animals used.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Patients baseline characteristics and the level of platinum in DRG of mice.
Additional file 2
: Raw data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, X., Jia, R., Hu, F. et al. Promoting AMPK/SR-A1-mediated clearance of HMGB1 attenuates chemotherapy-induced peripheral neuropathy. Cell Commun Signal 21, 99 (2023). https://doi.org/10.1186/s12964-023-01100-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01100-9