
Abstract
Liver fibrosis could be the last hope for treating liver cancer and remodeling of the hepatic microenvironment has emerged as a strategy to promote the ablation of liver fibrosis. In recent years, especially with the rapid development of nanomedicine, hepatic microenvironment therapy has been widely researched in studies concerning liver cancer and fibrosis. In this comprehensive review, we summarized recent advances in nano therapy-based remodeling of the hepatic microenvironment. Firstly, we discussed novel strategies for regulatory immune suppression caused by capillarization of liver sinusoidal endothelial cells (LSECs) and macrophage polarization. Furthermore, metabolic reprogramming and extracellular matrix (ECM) deposition are caused by the activation of hepatic stellate cells (HSCs). In addition, recent advances in ROS, hypoxia, and impaired vascular remodeling in the hepatic fibrotic microenvironment due to ECM deposition have also been summarized. Finally, emerging nanotherapeutic approaches based on correlated signals were discussed in this review. We have proposed novel strategies such as engineered nanotherapeutics targeting antigen-presenting cells (APCs) or direct targeting T cells in liver fibrotic immunotherapy to be used in preventing liver fibrosis. In summary, this comprehensive review illustrated the opportunities in drug targeting and nanomedicine, and the current challenges to be addressed.
Graphical Abstract

Similar content being viewed by others

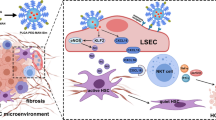
Introduction
Chronic liver disease (CLD) is a continuous and progressive pathological condition that progresses from initial fat accumulation to hepatitis and liver fibrosis, with end-stage fibrosis known as cirrhosis, which is highly susceptible to hepatocellular carcinoma (HCC) [1]. Approximately 12% of the global population (800 million people) suffer from liver disease and causing 2 million deaths each year [2]. Cancer is a worldwide threat to public health and HCC is the second leading cause of cancer death [3]. Is cancer curable? Almost all CLDs have provided convincing evidence confirming the reversibility of liver fibrosis and in clinical trials, in patients with chronic viral hepatitis and patients with advanced disease, timely "braking" interventions can subside fibrosis, which may be the last straw in the treatment of cancer [4,5,6].
The liver is highly complex and consists of two different cell entities, parenchymal cells, and non-parenchymal cells. Parenchymal cells include hepatocytes (60–70%) and cholangiocytes and account for most of the metabolic liver functions. While the non-parenchymal cells constitute 30–40% of total liver cells, like LSECs, HSCs, KCs, and other immune cells, playing a central role in the physiological processes of the liver [7]. The liver microenvironment, which fosters the survival and activity of liver cells, plays an important role in maintaining the normal structure and physiological function of the liver. The homeostasis of the liver microenvironment is disrupted during liver fibrosis development, causing hepatocyte damage, LSECs capillarization, HSCs activation, macrophage polarization, and immune cell suppression, and changing the cell–cell and cell–matrix interactions, which eventually form the hepatic fibrotic microenvironment [7, 8]. Recent studies have shown that modest prognostic performance (area under the receiver operating characteristic curve from 0.54 to 0.71) of five indirect markers of fibrosis (aspartate aminotransferase [AST]–to-platelet ratio index [APRI], Fibrosis-4 Index [FIB-4], BARD, Forns, NAFLD score [NAS]) [9] and direct markers-Liver Fibrosis test (LF) [10] to predict future development of cirrhosis and severe liver disease in the general population. And liver fibrosis can be assessed with relatively high accuracy noninvasively by serological tests, transient elastography, and radiological methods. These modalities may be utilized for screening for liver fibrosis in at-risk populations [11]. Nanoparticle (NPs) delivery systems have been widely studied as a drug delivery strategy in drug research and the liver targeting of NPs may be some of the nanomaterials beneficial for liver disease therapy [12], as shown in Fig. 1. NPs have diverse compositions, such as metallic NPs including metal (Ag and Au nanoparticles) and metal oxide (MOx) NPs including transition-metal oxides (TMOs, e.g., SiO2, ZnO, and TiO2), carbon NPs including 1D carbon nanotubes (CNTs) and 2D graphene-based NPs (FPL, and GO), cellulose nanocrystal (CNC) and cellulose nanofiber (CNF), Fluorescent NPs (quantum dots, CDs, Organic fluorophores), and organic NPs including lipid NPs, liposomes, and polymer NPs.

The major intrinsic properties of NPs and cellular uptake by the major liver cells
Although these NPs have a challenging and lengthy process, such as systemic circulation, drugs accumulation in the lesion, deep penetration, and intracellular release of drugs, the liver is one of the major aggregation organs for NPs, conventional NPs are recognized as foreign bodies, and rapidly captured by the reticuloendothelial system (RES) through the regulation of plasma proteins after administration, but the liver targeting NPs may serve as a beneficial nanomaterial for liver disease therapy [12], as shown in Fig. 2. As NPs move along the sinusoid, they will come into contact with sinusoidal endothelial cells, Kupffer cells (KCs), T cells, and DCs. Depending on their physicochemical properties, NPs have better access through fenestrae to enter the space of Disse and contact with hepatocytes. The smaller NPs may transcytose through the hepatocytes and enter the bile duct through bile canaliculi. Conventional NPs are captured by the RES. Larger size, negatively charged, or hydrophilic NPs are preferentially swallowed by KCs via phagocytosis; NPs less than 200 nm NP or with negative surface charge or hydrophobicity tend to be taken up by endothelial cells through clathrin-mediated endocytosis with a high exposure dose or long time. NPs less than 50 nm NP or hydrophilic NPs could be captured by stellate cells. Smaller NPs with positive surface charge or hydrophobic NPs are preferentially taken up by hepatocytes through clathrin-mediated endocytosis.

Interactions of NPs uptake and elimination in the liver during systematic circulation after NPs exposure
Hence, we reviewed the latest advances in remodeling the hepatic fibrotic microenvironment with emerging monotherapies, with particular attention to remodeling immune regulation, metabolic reprogramming, ECM deposition, and hypoxia-induced vascular production in the hepatic fibrotic microenvironment. Finally, the nano-advantages and challenges of engineered NPs targeting APCs or directly targeting T cells for immunotherapy of liver fibrosis were highlighted in this review.
Engineered NPs regulated immunosuppression-associated microenvironment
The immune system is a complex network including lymphoid organs, cells, and cytokines [13]. RES uptake and immune cell suppression are considered the main immune-related effects. In the liver, the RES, composed of LSECs and KCs, is the primary site of exposure to microbial antigens and plays a vital role in the uptake and clearance of soluble antigens from the hepatic sinusoids, serving as a guard against microbial invasion and maintenance of hepatic homeostasis [14]. Once fibrosis starts, LSECs change their phenotype (from fenestrations to capillaries) and then they open windows and the surface gradually shrinks to form an organized basement membrane, that is, "hepatic sinusoidal capillarization". In addition, LSECs serve as gatekeepers of the hepatic microenvironment and as platforms for innate or adaptive immune cells to stay in the hepatic sinusoidal microenvironment. This is important for maintaining systemic immune homeostasis [15].
LSECs-related immunosuppression-associated microenvironment
In the innate immune response, LSECs have an effective function by directly participating in the suppression of activated CD4 + T cells [16], or by expressing major histocompatibility complex (MHC) class I and II molecules presented to CD8 + T cells and promoting the activation of regulatory T cells (Tregs) [17]. In addition, LSECs express a variety of pattern recognition receptors including the toll-like receptor (TLR) family, scavenger receptors (SR-A, SR-B, and SR-H), and mannose receptors (MR) that produce inhibitory acquired immune responses. LSECs regulate adaptive immune responses directly by presenting antigens to T cells and also regulate natural killer T cells (NKT cells) by expressing CXCL16, and the cell surface ligand for CXCR6 [18]. Hepatic macrophages account for 90% of the total macrophages in the human body, and they are very plastic and adapt their phenotype according to signals derived from the hepatic fibrotic microenvironment [19]. They are divided into infiltrating macrophages and liver-resident macrophages[20]. Liver-resident macrophages, called KCs are self-renewed, resident, and non-migratory [19]. Liver injury triggers KCs activation, leading to inflammatory cytokine and chemokine release. This fosters the infiltration of monocytes into the liver, which gives rise to the large number of inflammatory monocyte-derived macrophages [21] Moreover, KCs promote T cell-mediated hepatitis development by producing CXCL10 and limiting the permeability of hepatic LSECs [22]. The macrophage pool of the liver can be rapidly expanded by infiltrating phagocytes that mainly originate from peripheral blood marrow/monocyte-derived macrophages [21], a few from peritoneal macrophages [23], and splenic macrophages [24]. In mice, two major populations of circulating monocytes exist Ly-6C high (Ly-6Chi) and Ly-6C low (Ly-6Clo) expressing monocytes. Whereas the Ly-6Chi monocytes express inflammatory chemokine receptors (like CCR2), pattern-recognition receptors, and cytokines [25]. The bone marrow is the primary source of the (relatively immature) Ly-6Chi monocytes [26], whereas the spleen serves as a reservoir for Ly-6Clo monocytes, the production of TNF-α, IL-6, and IL-10 increased significantly in hepatic splenic macrophages and migrate from bone marrow to the liver via the spleen [27]. As a consequence of tissue injury, KCs and other liver cells (HSCs, hepatocytes) secrete chemokines like CCL2 that provoke the massive infiltration of Ly6Chi monocytes into the injured liver [28]. This provides a rapid and transient mechanism to expand the macrophage pool in the liver by inflammation-prone phagocytes. Together, they form a "profibrogenic environment" in which immune homeostasis is disrupted, which may explain the refractory nature of immunotherapy for liver fibrosis, as shown in Table 1.
Polymeric micelles (PMs) as drug delivery vehicles have specific targeting and high stability in vivo as illustrated in Fig. 3. Scavenger receptors form a superfamily of membrane-bound receptors, and in vitro studies have shown that anionic NPs modified with scavenger receptor stabilizer-2 (stab2) receptor ligands target naturally tolerant LSECs and generate Tregs, thereby suppressing antigen-specific immune responses [29] and which also leads to selective deletion of single blood vessels in zebrafish embryos, with important clearance of lipoprotein B-containing lipoproteins in zebrafish [30]. Hyaluronic acid (HA) is a naturally occurring ligand and encapsulated micelles that show sustained drug release and low cytotoxicity for targeting LSECs, with over 90% of HA in the blood that is absorbed and metabolized by LSECs [31] LSEC-targeting and fenestrae-repairing nanoparticles (named HA-NPs/SMV) rapidly released SMV and exerted a fenestrae-repairing function, providing an antifibrotic therapeutic regimen. [32]. Studies have shown that coating positively charged PMs consisting of poly(L-lysine)-block-poly(L-lactide) (PLys-b-PLLA) AB diblock copolymers with anionic HA by polyion complex (PIC) formation target specific interaction between LSECs and KCs, thereby increasing the toxic T-lymphocyte/Treg cell ratio and then cause immune inflammation [33]. Poly (lactic-co-glycolic acid) (PLGA) has been used in many long-acting drug formulations approved by the US Food and Drug Administration (FDA) [34]. NPs decorated with stab ligands and PLGA target naturally tolerant LSECs are capable of producing Tregs to inhibit antigen-specific immune responses [33].

The NPs in LSECs capillarization-mediated immunosuppression-associated microenvironment of liver fibrosis
Macrophages-related immunosuppression-associated microenvironment
Activated macrophages can release cytokines, which can affect the adaptive immune response [35]. It was illustrated in Fig. 4 that MR directly promotes the pro-inflammatory activation of macrophages and triggers inflammation [36]. Mannose-modified albumin NPs have the potential to deliver TGFβ-siRNA to CD206 + macrophages as an anti-fibrotic strategy [37]. A stable nucleic acid–lipid particle delivery system of mannose-modified HMGB1-siRNA (mLNP-siHMGB1) targets hepatic macrophages via mannose receptor-mediated targeting, thereby silencing HMGB1 protein expression and inhibits the activation of HSCs for the treatment of liver fibrosis [38]. Mannose-modified trimethyl chitosan-cysteine (MTC)-coupled NPs as efficient polymeric carriers for oral TNF-α siRNA, and a dextran-based siRNA carrier system, BG34-10-Re-I/siRNA, was also developed for macrophage-targeted siRNA delivery [39]. Carboxymethyl chitosan (CMC) is a promising drug-release polymeric carrier with biocompatible, biodegradable, and easily accessible features. CMC reduces host immune response in an experimental mouse model of CCl4-induced chronic liver injury [40]. Phosphatidylserine (PS) is a phospholipid with a negatively charged head that is normally found in the inner leaflet of the cell membrane. PS-containing NPs are commonly used to mimic apoptotic cells and can specifically modulate macrophage function and enhance the targeting ability of macrophages [41], PS-modified nanostructured lipid carriers were further designed in some previous studies to improve hepatic delivery efficiency and its bioavailability, thereby reducing liver fibrosis and collagen fiber deposition in vivo [42]. The scaffolds crosslinked with nano-graphene oxide show high resistance to enzymatic degradation via direct inhibition of MMPs activity and increased M2-like macrophage polarization, which reduces graft-elicited inflammation (Table 2). Overall, nano-graphene oxide offers an alternative for donor organs [43].

The NPs in macrophages polarization-mediated immunosuppression-associated microenvironment of liver fibrosis
Engineered NPs regulated metabolic reprogramming-associated microenvironment
Liver injury stimulates the transdifferentiation of quiescent hepatic stellate cell HSCs to proliferative, migratory, and fibrotic myofibroblastic (MFs) [44]. The short-term accumulation of MFs is beneficial for liver regeneration, however, long-term excessive accumulation results in progressive fibrosis, repair defects, and increase risk and mortality from cirrhosis and liver cancer [45]. The acquisition of MFs phenotype is energy-intensive, whereas reprogramming of HSCs in metabolism is similar to that of highly proliferative cancer cells, and orchestrating the reprogramming of HSCs is a novel therapeutic target for fibrosis therapy [46, 47], including.
-
(1)
Enhanced aerobic glycolysis [48]. Elevation in glycolysis is related to an increase in glucose transporter proteins, including GLUT1, which is highly overexpressed in cancer cells [49] In addition, an increase in glycolysis is accompanied by a shunt of central carbon metabolites from the citric acid cycle, including increased expression of pyruvate dehydrogenase kinase 3 (PDK3), which promotes lactate production. A similar phenomenon is called the Warburg effect in cancer cells [50]. Furthermore, pyruvate kinase M2 (PKM2) represents a unique link between aHSCs and cancer cells that promote aerobic glycolysis [51].
-
(2)
Upregulation of glutamine catabolism [52]. Glutamine metabolism has been identified as an additional source of ATP in HSCs. Liver samples from patients with nonalcoholic steatohepatitis (NASH) and advanced fibrosis [53] and a mouse model of liver fibrosis [54] have shown that the proliferation of HSCs is heavily dependent on glutamine metabolism [52]. Metabotropic glutamate receptor-5 (mGluR5) production of 2-arachidonic acid glycerol (2-AG) in HSCs activates hepatocyte cannabinoid receptor-1 (CB1R)-mediated neoadipogenesis [55].
-
(3)
Fatty acid catabolism [56]. Compared to the resting HSCs, activated HSCs have abundant mitochondria, and the requirement to maintain the phenotype of MFs still needs the energetic contribution of oxidative phosphorylation (OXPHOS). It has been shown that mitochondrial uncoupling inhibits the activation of HSCs in vitro, despite increased glycolysis [57]. The metabolism of lipid droplets that provide fatty acids for mitochondrial β-oxidation (FAO) plays an important role in lipid metabolic pathways [58]. Similarly, in cancer, FAO survives oxidative stress and nutrient deprivation [56].
HSCs-related metabolic reprogramming-associated microenvironment
It was illustrated in Fig. 5 that the HSCs expressed the mannose-6-phosphate/insulin-like growth factor II (M6P/IGFII) receptor, and M6P-modified albumin activates HSCs in the fibrotic liver, blocks glutamine (GLN) catabolism by mediating the Hedgehog signaling pathway Glutaminases including GLS (glutaminase), aspartate aminotransferase (GOT1) and glutamate dehydrogenase (GLUD1) and inhibits myofibrillar activity [59]. GLN metabolism is an important component of metabolic reprogramming. GLN can be converted to α-ketoglutarate (α-KG) to provide carbon for the TCA cycle, or to other NEAAs via transaminases (GOT1 and GOT2). GLN can also be converted to glutamate and pyrroline-5-carboxylic acid (P5C) to stimulate collagen biosynthesis [60]. HSCs have multiple vitamin A (VA)-rich lipid droplets in the cytoplasm, which are the primary sites for retinoid derivatives storage in vivo. A study has synthesized a novel VA-Myrj52 ester conjugated solid lipid NPs (VA-SLNs) using all-trans retinoic acid and hydrophilic emulsifier (Myrj52) as targeting agents to effectively reduce peroxisome proliferator-activated receptor γ (PPARγ)/SREBPs-mediated lipid accumulation [61]. SREBPs are highly expressed to promote tumor growth, especially in the regulation of lipid metabolism [62]. In addition, chondroitin sulfate PMs target HSCs in liver fibrosis [63]. Green biosynthesis of NPs using reduced metabolites of microbial and plant-derived products is a better strategy for achieving inexpensive products that are less harmful to health and the environment compared to artificial physical or chemically manufactured NPs [64]. TGFβ signaling is involved in stimulating glycolysis and mitochondrial respiration. Inhibitory effect of curcumin/chitosan-coated green silver NPs directly bound to the fibrogenic protein TGF-β [65]. Mouse livers were decellularized to form liver hydrogels as an injectable biomaterial in the liver, which blocked the TGF-β1/Smad pathway to reduce fibrosis [66] (Table 2).

The NPs in HSCs activation-mediated metabolic reprogramming-associated microenvironment of liver fibrosis
Immune cells related metabolic reprogramming-associated microenvironment
Regarding macrophages and KCs, restriction of their glucose and glutamine supply inhibits their secretory function [67]. Conversely, altered lipid metabolism causes KCs to accumulate cytotoxic lipids to enhance the proinflammatory phenotype [68]. The enhanced glycolytic activity, altered tricarboxylic acid cycle, and reduced ATP production in the macrophages under 100–125 nm diameter NPs made of silk, poly(lactic-ethanolic acid), or silicon consistent with a pro-inflammatory phenotype [69]. PEGylation of cellulose nanofiber (CNF) reduces macrophage-initiated inflammatory and metabolic responses, including increased glycolysis and reprogramming of the tricarboxylic acid cycle and the creatine kinase/phosphocreatine pathway [70]. Energy metabolism is also involved in the body's immune network. It has been shown that metabolic reprogramming between immune cells and cancer-associated fibroblasts (CAFs) indicated the immune status of tumors [71]. aCD3/F/AN-induced lipid metabolic reprogramming specifically activates T cells [72]. A recent study synthesizes cholesterol-modified polymeric CXCR4 inhibitor (Chol-PCX) in the form of Chol-PCX/miRNA NPs and CXCL12/CXCR4 axis disrupts the lipid metabolic network of T cells for the amplified treatment of liver fibrosis [73, 74], suggesting that nanotechnology-enabled T cell lipid metabolic reprogramming has the potential to be a new paradigm for immunometabolic therapy.
Engineered NPs regulated hypoxia-associated microenvironment
Activated HSCs lose the ability to store retinol, then they begin to proliferate and produce pro-fibrotic cytokines such asα-smooth muscle actin (α-SMA), type I, and type III collagens [44]. Whereas collagen fibers between adjacent hepatic blood sinusoids contact each other and encircle hepatocytes in a grid pattern, which weakens oxygen exchange between hepatic sinusoids and hepatocytes, therefore leads to hypoxia in the microenvironment of liver fibrosis [75]. In addition, abnormal immune inflammation leads to the release of inflammatory factors, chemokines, ROS, adipokines, and pro-angiogenic mediators [76]. Among them, intracellular NO and ROS reactions are significantly increased in extracellular matrix (ECM) subjected to peroxynitrite (ONOO-)-mediated oxidization, and MMP-2 activity, directly mediated by S-glutathionylation of its cysteine residues in the presence of ONOO(-) and by phosphorylation of its serine and threonine residues [77], as well as increased fibrosis [78]. Excessive deposition of fibrillated collagen (mainly collagen I), the main collagen of ECM, in the Disse space would greatly impede the delivery of HSCs by nanoformulations [157]. However, there are few relevant studies and in-depth studies are needed in the future.
Conclusions and outlooks
Engineered NPs are ideal tools for the application of liver fibrosis therapy. However, only a few have been successfully translated into clinical practice, mainly due to the macrophage uptake of NPs. Disulfide bonds are commonly used as intermediate linkers in the fabrication of silicon networks, and disulfide-bonded organosilicon NPs with cage-like morphology target LSECs to avoid macrophage filtration and may affect the tolerance status of intrahepatic immune cells [158]. (1) Targeting specific cell types is expected to be a useful technique, suggesting the use of nanomedicines for diagnostic, therapeutic, and prognostic integration of personalized treatment of liver fibrosis is an important future drug development direction. LNPs accumulate in the LSECs to cause activation and neutrophil inflammation. Furthermore, the modification of N-acetyl-D-galactosamine (GalNAc)-LNPs with polyethylene glycol eliminates the toxicity associated with LNPs [159]. (2) Combination therapies with different mechanisms can be used for the development of engineered nanotherapeutics. The microenvironment of liver fibrosis is the result of multiple cellular interactions upon encountering the capillaries LSEC barrier, HA-NPs/SMV rapidly released SMV and exerted a fenestrae-repairing function, which allowed more CV-NPs/siCol1α1 to enter the space of Disse to degrade deposited collagen and finally to achieve higher accumulation in activated HSCs, promoting pathological barrier-normalization [32]. (3) The biosafety of the NPs should be investigated more. Although the biocompatibility of the NPs have been confirmed in vitro or in vivo. But when they need to be transferred to the clinical trials, such as the biodistribution after the injection, or the biodegradation pathway in different organs.
In addition to traditional drug therapies, nano vaccines allow precise modulation of the composition and structure of NPs, their physicochemical properties (size, shape, function, and surface charge) as well as the dose and route of administration of nano drugs, to improve antigen presentation and strong immunogenicity. Therefore, it has become an attractive alternative or complementary therapy in the field of cancer treatment [160] and has received lots of attention in the treatment of liver fibrosis. Polystyrene NPs, PLGA, CNTs, aluminum hydroxide NPs, SiO2 NPs, carbon black NPs, and TiO2 NPs, have been shown to stimulate NLRP3-related inflammasomes [161], and liposomes, polymers, and inorganic NPs, as well as self-assembled protein NPs and virus-like particles (VLPs), are being explored as antigen carriers [160]. Furthermore, achieving subcellular localization of drug loading is critical to maximizing the therapeutic potential of a drug [162]. Carbon dots (CDs) are new fluorescent nanomaterials with negligible photobleaching, among which graphene quantum dots (GQDs) can be used for super-selective cell nuclear imaging due to their superior biocompatibility and targeting ability [163]. In addition, quantum dots (QDs) are used for imaging and drug-targeted delivery, which allow rapid absorption in the small intestine after oral administration and is highly specific targeting to LSECs or hepatocytes [164]. Moreover, graphene oxide (GO) enables efficient liver regeneration via immunomodulation [43].
Meanwhile, the hepatic fibrosis microenvironment, composed of immune cells, MFs, LSECs, HSCs, and ECM with abundant growth/signaling factors, is a unique, complex, and highly dynamic region. These diverse cell types and ECM proteins are capable of coordinating liver remodeling, hematopoiesis, regulation of immune function, and tissue regeneration. Reprogramming the tumor microenvironment (TIME) and reversing immunosuppressive strategies are currently the most beneficial modalities for cancer therapy [165]. In addition, the hepatic fibrosis microenvironment and reversal of immunosuppression are the most promising strategies for liver fibrosis treatment. Moreover, the energy utilization of immune cells differs significantly in T cells exhibiting completely different metabolic patterns depending on the activation state [166]. For example, the metabolism of naive T cells is essentially static [167], exhibiting zero proliferation, therefore, only minimal nutrient intake. Minimal glycolytic rates and minimal biosynthesis are required to maintain reliance on OXPHOS to provide ATP [168]. As it becomes metabolically activated, there is an increased nutrient uptake, enhanced glycolysis rate, synthesis, and accumulation of proteins, lipids, and nucleotides, as well as the growth and proliferation of T cells to perform killing functions [168]. Memory T cells have a similar metabolic pattern to naive T cells, maintaining a basic nutrient intake, a lower glycolytic rate, and relying on OXPHOS for ATP [169]. In addition, activated NK cells [170], neutrophils [171], M1 macrophages [172], and DCs [173] rely mainly on glycolysis for energy supply. However, DCs used mainly oxidative phosphorylation for energy metabolism at resting state. In addition, aerobic glycolysis and pentose phosphate pathway are the main metabolic modes of neutrophils [174]. More interestingly, glycolysis and mitochondrial metabolism are enhanced following the activation of B lymphocytes induced by LPS stimulation or antigenic stimulation. However, glycolysis is the main metabolism in activated B lymphocytes [175]. However, Treg cells [176] and M2 macrophages [177] mainly rely on OXPHOS (FAO) from fatty acid oxidation for energy supply. It is suggested that novel immune cell metabolic reprogramming targeting biodegradability, specific selectivity, responsive drug release, and multimodal synergistic therapy of engineered nanomaterials have broad applications in the treatment of liver fibrosis.
Nanomedicine for fibrosis offers the opportunity to enhance the anti-fibrotic immune response, achieve specificity and local amplification of the immune response safely and effectively in fibrotic tissue, and improve the rate of patients` treatment for immunotherapy, as well as reduce related side effects. Several nanoparticle-based T-cell therapies remained unexploited. For example, more studies in recent years have identified new subpopulations of immune cells [178] and these subgroups are considered to be homogeneous groups, which may be a new target for immunotherapy, and the mechanisms of these therapies need to be further understood. The ability to manufacture "generic" or "off-the-shelf" NPs using antigen diagnostics will reduce the cost burden and expand the range of patients to be effectively treated with T-cell immunotherapy compared to cell-based therapies. In addition, multi-reactive nanomedicine for immunotherapy comprehensively modulates complex pathogenic processes. Therefore, NP-mediated T-cell immunotherapy will have sustained progress in the future.
Data availability
Not applicable.
Change history
12 May 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12951-023-01914-2
Abbreviations
- APCs:
-
Antigen-presenting cells
- APRI:
-
AST-to-platelet ratio index
- ASO:
-
Antisense oligonucleotides
- AST:
-
Aspartate aminotransferase
- CAFs:
-
Cancer-associated fibroblasts
- CDs:
-
Carbon dots
- CB1R:
-
Cannabinoid receptor-1
- ChiBil:
-
Chitosan-bilirubin micelle
- CLD:
-
Chronic liver disease
- CLRs:
-
C-type lectin receptors
- CMC:
-
Carboxymethyl chitosan
- CNF:
-
Cellulose nanofiber
- ConA:
-
Concanavalin A
- COVID-19:
-
Coronavirus pneumonia
- ECM:
-
Extracellular matrix
- EO:
-
Ethylene oxide
- FAO:
-
Fatty acids for mitochondrial β-oxidation
- FDA:
-
Food and Drug Administration
- FIB-4:
-
Fibrosis-4 index
- FPL:
-
Fulleropyrrolidine
- GLN:
-
Glutamine
- GLUD1:
-
Glutamate dehydrogenase
- GO:
-
Graphene oxide
- GOT1:
-
Aspartate aminotransferase
- GQDs:
-
Graphene quantum dots
- HA:
-
Hematoxylic acid
- HCC:
-
Hepatocellular carcinoma
- IHVR:
-
Intrahepatic vascular resistance
- IL:
-
Interleukin-10
- KCs:
-
Kupffer cells
- LF:
-
Liver Fibrosis test
- LSECs:
-
Liver sinusoidal endothelial cells
- LNPs:
-
Lipid nanoparticles
- MFs:
-
Myofibroblastic
- mGluR5:
-
Metabotropic glutamate receptor-5
- MHC:
-
Major histocompatibility complex
- mLNP-siHMGB1:
-
Mannose-modified HMGB1-siRNA
- MR:
-
Mannose receptors
- MTC:
-
Mannose-modified trimethyl chitosan-cysteine
- M6P/IGFII:
-
Mannose-6-phosphate/insulin-like growth factor II
- NASH:
-
Nonalcoholic steatohepatitis
- NAS:
-
NAFLD score
- NKT cells:
-
Natural killer T cells
- NO:
-
Nitric oxide
- NPs:
-
Nanoparticle
- OXPHOS:
-
Oxidative phosphorylation
- PDK3:
-
Pyruvate dehydrogenase kinase 3
- PEI:
-
Polyethyleneimine
- PEG:
-
Polyethylene glycol
- PVA:
-
Polyvinyl alcohol
- PKM2:
-
Pyruvate kinase M2
- PLGA:
-
Poly (lactic-co-glycolic acid)
- PMs:
-
Polymeric micelles
- PO:
-
Propylene oxide
- pro-MMPs:
-
Pro-matrix metalloproteinases
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PS:
-
Phosphatidylserine
- P5C:
-
Pyrroline-5-carboxylic acid
- QDs:
-
Quantum dots
- RAP:
-
Allowing RcP carriers
- RBP4:
-
Particularly retinol-binding protein 4
- RcP:
-
Further binds to nucleotides
- RES:
-
Reticuloendothelial system
- RNAi:
-
RNA interference
- SLNs:
-
Solid lipid NPs
- SPION:
-
Suoxide pellets
- SR:
-
Scavenger receptors
- stab2:
-
Scavenger receptor stabilizer-2
- TIME:
-
Tumor microenvironment
- TLR:
-
Toll-like receptor
- Tregs:
-
Regulatory T cells
- USSN:
-
Ultrasmall silica NPs
- VLPs:
-
Virus-like particles
- VA:
-
Vitamin A
- 2-AG:
-
2-Arachidonic acid glycerol
- α-KG:
-
α-Ketoglutarate
References
Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet. 2021;398(10308):1359–76.
Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70(1):151–71.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Dhar D, Baglieri J, Kisseleva T, Brenner DA. Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med. 2020;245(2):96–108.
Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4):875.
Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: concept to treatment. J Hepatol. 2015;62(1 Suppl):S15-24.
Zhao X, Xue X, Cui Z, Kwame Amevor F, Wan Y, Fu K, Wang C, Peng C, Li Y. microRNAs-based diagnostic and therapeutic applications in liver fibrosis. Wiley Interdiscip Rev RNA. 2022. https://doi.org/10.1002/wrna.1773.
Meng Y, Zhao T, Zhang Z, Zhang D. The role of hepatic microenvironment in hepatic fibrosis development. Ann Med. 2022;54(1):2830–44.
Hagström H, Talbäck M, Andreasson A, Walldius G, Hammar N. Ability of noninvasive scoring systems to identify individuals in the population at risk for severe liver disease. Gastroenterology. 2020;158(1):200–14.
Thiele M, Madsen BS, Hansen JF, Detlefsen S, Antonsen S, Krag A. Accuracy of the enhanced liver fibrosis test vs fibrotest, elastography, and indirect markers in detection of advanced fibrosis in patients with alcoholic liver disease. Gastroenterology. 2018;154(5):1369–79.
Ginès P, Castera L, Lammert F, Graupera I, Serra-Burriel M, Allen AM, Wong VW, Hartmann P, Thiele M, Caballeria L, de Knegt RJ, Grgurevic I, Augustin S, Tsochatzis EA, Schattenberg JM, Guha IN, Martini A, Morillas RM, Garcia-Retortillo M, de Koning HJ, Fabrellas N, Pich J, Ma AT, Diaz MA, Roulot D, Newsome PN, Manns M, Kamath PS, Krag A. Population screening for liver fibrosis: toward early diagnosis and intervention for chronic liver diseases. Hepatology. 2022;75(1):219–28.
Li J, Chen C, **a T. Understanding nanomaterial-liver interactions to facilitate the development of safer nanoapplications. Adv Mater. 2022;34(11):e2106456.
Parkin J, Cohen B. An overview of the immune system. Lancet. 2001;357(9270):1777–89.
Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou PE. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2017;66(1):212–27.
Wohlleber D, Knolle PA. The role of liver sinusoidal cells in local hepatic immune surveillance. Clin Transl Immunology. 2016;5(12): e117.
Tang L, Yang J, Liu W, Tang X, Chen J, Zhao D, Wang M, Xu F, Lu Y, Liu B, Sun Q, Zhang L, He F. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology. 2009;137(4):1498-508.e1-5.
Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, Arnold B, Knolle PA. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6(12):1348–54.
Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3(4): e113.
Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–12.
Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, Zhou H, Li Y. Macrophage polarization and its role in liver disease. Front Immunol. 2021;12: 803037.
Watanabe Y, Tsuchiya A, Seino S, Kawata Y, Kojima Y, Ikarashi S, Starkey Lewis PJ, Lu WY, Kikuta J, Kawai H, Yamagiwa S, Forbes SJ, Ishii M, Terai S. Mesenchymal stem cells and induced bone marrow-derived macrophages synergistically improve liver fibrosis in mice. Stem cells translational medicine. 2019;8(3):271–84.
Dai S, Liu F, Qin Z, Zhang J, Chen J, Ding WX, Feng D, Ji Y, Qin X. Kupffer cells promote T-cell hepatitis by producing CXCL10 and limiting liver sinusoidal endothelial cell permeability. Theranostics. 2020;10(16):7163–77.
García-Peñarrubia P, Ruiz-Alcaraz AJ, Ruiz-Ballester M, Ramírez-Pávez TN, Martínez-Esparza M. Recent insights into the characteristics and role of peritoneal macrophages from ascites of cirrhotic patients. World J Gastroenterol. 2021;27(41):7014–24.
Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. 2022;55(9):1515–29.
Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, Heymann F, Kalthoff S, Lefebvre E, Eulberg D, Luedde T, Marx G, Strassburg CP, Roskams T, Trautwein C, Tacke F. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64(5):1667–82.
Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7(3):311–7.
Fukushima H, Kono H, Hirayama K, Akazawa Y, Nakata Y, Wakana H, Fujii H. Changes in function and dynamics in hepatic and splenic macrophages in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2020;13:305–14.
Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50(1):261–74.
Campbell F, Bos FL, Sieber S, Arias-Alpizar G, Koch BE, Huwyler J, Kros A, Bussmann J. Directing nanoparticle biodistribution through evasion and exploitation of stab2-dependent nanoparticle uptake. ACS Nano. 2018;12(3):2138–50.
Verwilligen RAF, Mulder L, Rodenburg FJ, Van Dijke A, Hoekstra M, Bussmann J, Van Eck M. Stabilin 1 and 2 are important regulators for cellular uptake of apolipoprotein B-containing lipoproteins in zebrafish. Atherosclerosis. 2022;346:18–25.
DeLeve LD, Maretti-Mira AC. Liver sinusoidal endothelial cell: an update. Semin Liver Dis. 2017;37(4):377–87.
Zhang LF, Wang XH, Zhang CL, Lee J, Duan BW, **ng L, Li L, Oh YK, Jiang HL. Sequential nano-penetrators of capillarized liver sinusoids and extracellular matrix barriers for liver fibrosis therapy. ACS Nano. 2022;16(9):14029–42.
Ohya Y, Takeda S, Shibata Y, Ouchi T, Kano A, Iwata T, Mochizuki S, Taniwaki Y, Maruyama A. Evaluation of polyanion-coated biodegradable polymeric micelles as drug delivery vehicles. J Control Release. 2011;155(1):104–10.
Wang Y, Qin B, **a G, Choi SH. FDA’s poly (lactic-co-glycolic acid) research program and regulatory outcomes. AAPS J. 2021;23(4):92.
Binatti E, Gerussi A, Barisani D, Invernizzi P. The role of macrophages in liver fibrosis: new therapeutic opportunities. Int J Mol Sci. 2022;23(12):6649.
van der Zande HJP, Nitsche D, Schlautmann L, Guigas B, Burgdorf S. The mannose receptor: from endocytic receptor and biomarker to regulator of (meta) inflammation. Front Immunol. 2021;12: 765034.
Singh A, Chakraborty S, Wong SW, Hefner NA, Stuart A, Qadir AS, Mukhopadhyay A, Bachmaier K, Shin JW, Rehman J, Malik AB. Nanoparticle targeting of de novo profibrotic macrophages mitigates lung fibrosis. Proc Natl Acad Sci USA. 2022;119(15): e2121098119.
Zhou JE, Sun L, Liu L, Jia Y, Han Y, Shao J, Wang J, Wang Y, Yu L, Yan Z. Hepatic macrophage targeted siRNA lipid nanoparticles treat non-alcoholic steatohepatitis. J Control Release. 2022;343:175–86.
Zhang M, Gao Y, Caja K, Zhao B, Kim JA. Non-viral nanoparticle delivers small interfering RNA to macrophages in vitro and in vivo. PLoS ONE. 2015;10(3): e0118472.
Gou Y, Weng Y, Chen Q, Wu J, Wang H, Zhong J, Bi Y, Cao D, Zhao P, Dong X, Guo M, Wagstaff W, Hendren-Santiago B, Chen C, Youssef A, Haydon RC, Luu HH, Reid RR, Shen L, He TC, Fan J. Carboxymethyl chitosan prolongs adenovirus-mediated expression of IL-10 and ameliorates hepatic fibrosis in a mouse model. Bioeng Transl Med. 2022;7(3): e10306.
Wang J, Kang YX, Pan W, Lei W, Feng B, Wang XJ. Enhancement of anti-inflammatory activity of curcumin using phosphatidylserine-containing nanoparticles in cultured macrophages. Int J Mol Sci. 2016;17(5):969.
Wang J, Pan W, Wang Y, Lei W, Feng B, Du C, Wang XJ. Enhanced efficacy of curcumin with phosphatidylserine-decorated nanoparticles in the treatment of hepatic fibrosis. Drug Delivery. 2018;25(1):1–11.
Kim DH, Kim MJ, Kwak SY, Jeong J, Choi D, Choi SW, Ryu J, Kang KS. Bioengineered liver crosslinked with nano-graphene oxide enables efficient liver regeneration via MMP suppression and immunomodulation. Nat Commun. 2023;14(1):801.
Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017;65(3):1039–43.
Ritz T, Krenkel O, Tacke F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol. 2018;330:175–82.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411.
Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42.
Gajendiran P, Vega LI, Itoh K, Sesaki H, Vakili MR, Lavasanifar A, Hong K, Mezey E, Ganapathy-Kanniappan S. Elevated mitochondrial activity distinguishes fibrogenic hepatic stellate cells and sensitizes for selective inhibition by mitotropic doxorubicin. J Cell Mol Med. 2018;22(4):2210–9.
Trivedi P, Wang S, Friedman SL. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33(2):242–57.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–8.
Zheng D, Jiang Y, Qu C, Yuan H, Hu K, He L, Chen P, Li J, Tu M, Lin L, Chen H, Lin Z, Lin W, Fan J, Cheng G, Hong J. Pyruvate kinase M2 tetramerization protects against hepatic stellate cell activation and liver fibrosis. Am J Pathol. 2020;190(11):2267–81.
Matés JM, Campos-Sandoval JA, Santos-Jiménez JL, Márquez J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019;467:29–39.
Li J, Ghazwani M, Liu K, Huang Y, Chang N, Fan J, He F, Li L, Bu S, **e W, Ma X, Li S. Regulation of hepatic stellate cell proliferation and activation by glutamine metabolism. PLoS ONE. 2017;12(8): e0182679.
Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, Diehl AM. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154(5):1465-1479.e13.
Choi WM, Kim HH, Kim MH, Cinar R, Yi HS, Eun HS, Kim SH, Choi YJ, Lee YS, Kim SY, Seo W, Lee JH, Shim YR, Kim YE, Yang K, Ryu T, Hwang JH, Lee CH, Choi HS, Gao B, Kim W, Kim SK, Kunos G, Jeong WI. Glutamate signaling in hepatic stellate cells drives alcoholic steatosis. Cell Metab. 2019;30(5):877-889.e7.
Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, Fang X. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100.
Guimarães EL, Best J, Dollé L, Najimi M, Sokal E, van Grunsven LA. Mitochondrial uncouplers inhibit hepatic stellate cell activation. BMC Gastroenterol. 2012;12:68.
Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–46.
Luk JM, Zhang QS, Lee NP, Wo JY, Leung PP, Liu LX, Hu MY, Cheung KF, Hui CK, Lau GK, Fan ST. Hepatic stellate cell-targeted delivery of M6P-HSA-glycyrrhetinic acid attenuates hepatic fibrogenesis in a bile duct ligation rat model. Liver Int. 2007;27(4):548–57.
Huang W, Choi W, Chen Y, Zhang Q, Deng H, He W, Shi Y. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23(5):724–7.
Mahdinloo S, Hemmati S, Valizadeh H, Mahmoudian M, Mahmoudi J, Roshangar L, Sarfraz M, Zakeri-Milani P. Synthesis and preparation of vitamin A coupled butein-loaded solid lipid nanoparticles for liver fibrosis therapy in rats. Int J Pharm. 2022;625: 122063.
Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38(1):27.
Luo J, Zhang P, Zhao T, Jia M, Yin P, Li W, Zhang ZR, Fu Y, Gong T. Golgi apparatus-targeted chondroitin-modified nanomicelles suppress hepatic stellate cell activation for the management of liver fibrosis. ACS Nano. 2019;13(4):3910–23.
Yadi M, Mostafavi E, Saleh B, Davaran S, Aliyeva I, Khalilov R, Nikzamir M, Nikzamir N, Akbarzadeh A, Panahi Y, Milani M. Current developments in green synthesis of metallic nanoparticles using plant extracts: a review. Artif Cells Nanomed Biotechnol. 2018;46(sup3):S336-s343.
Elzoheiry A, Ayad E, Omar N, Elbakry K, Hyder A. Anti-liver fibrosis activity of curcumin/chitosan-coated green silver nanoparticles. Sci Rep. 2022;12(1):18403.
Hussein KH, Park KM, Yu L, Kwak HH, Woo HM. Decellularized hepatic extracellular matrix hydrogel attenuates hepatic stellate cell activation and liver fibrosis. Mater Sci Eng C Mater Biol Appl. 2020;116: 111160.
Schwaderer J, Phan TS, Glöckner A, Delp J, Leist M, Brunner T, Delgado ME. Pharmacological LRH-1/Nr5a2 inhibition limits pro-inflammatory cytokine production in macrophages and associated experimental hepatitis. Cell Death Dis. 2020;11(2):154.
Bieghs V, Walenbergh SM, Hendrikx T, van Gorp PJ, Verheyen F, Olde Damink SW, Masclee AA, Koek GH, Hofker MH, Binder CJ, Shiri-Sverdlov R. Trap** of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int. 2013;33(7):1056–61.
Saborano R, Wongpinyochit T, Totten JD, Johnston BF, Seib FP, Duarte IF. Metabolic reprogramming of macrophages exposed to silk, poly(lactic-co-glycolic acid), and silica nanoparticles. Adv Healthc Mater. 2017. https://doi.org/10.1002/adhm.201601240.
Totten JD, Wongpinyochit T, Carrola J, Duarte IF, Seib FP. PEGylation-dependent metabolic rewiring of macrophages with silk fibroin nanoparticles. ACS Appl Mater Interfaces. 2019;11(16):14515–25.
Zhu Y, Li X, Wang L, Hong X, Yang J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front Endocrinol. 2022;13: 988295.
Kim D, Wu Y, Li Q, Oh YK. Nanoparticle-mediated lipid metabolic reprogramming of T cells in tumor microenvironments for immunometabolic therapy. Nano Micro Letters. 2021;13(1):31.
Zhang C, Hang Y, Tang W, Sil D, Jensen-Smith HC, Bennett RG, McVicker BL, Oupický D. Dually active polycation/miRNA nanoparticles for the treatment of fibrosis in alcohol-associated liver disease. Pharmaceutics. 2022;14(3):669.
Wu P, Luo X, Sun M, Sun B, Sun M. Synergetic regulation of kupffer cells, extracellular matrix and hepatic stellate cells with versatile CXCR4-inhibiting nanocomplex for magnified therapy in liver fibrosis. Biomaterials. 2022;284: 121492.
Couvelard A, Scoazec JY, Dauge MC, Bringuier AF, Potet F, Feldmann G. Structural and functional differentiation of sinusoidal endothelial cells during liver organogenesis in humans. Blood. 1996;87(11):4568–80.
Zhao X, Wang C, Dai S, Liu Y, Zhang F, Peng C, Li Y. Quercetin protects ethanol-induced hepatocyte pyroptosis via scavenging mitochondrial ROS and promoting PGC-1α-regulated mitochondrial homeostasis in L02 cells. Oxid Med Cell Longev. 2022;2022:4591134.
Krzywonos-Zawadzka A, Franczak A, Sawicki G, Bil-Lula I. Mixture of MMP-2 MLC, and NOS inhibitors affects NO metabolism and protects heart from cardiac I/R injury. Cardiol Res Pract. 2020;2020:1561478.
Yuen VW, Wong CC. Hypoxia-inducible factors and innate immunity in liver cancer. J Clin Investig. 2020;130(10):5052–62.
Fan QQ, Zhang CL, Qiao JB, Cui PF, **ng L, Oh YK, Jiang HL. Extracellular matrix-penetrating nanodrill micelles for liver fibrosis therapy. Biomaterials. 2020;230: 119616.
Zhou L, Liang Q, Li Y, Cao Y, Li J, Yang J, Liu J, Bi J, Liu Y. Collagenase-I decorated co-delivery micelles potentiate extracellular matrix degradation and hepatic stellate cell targeting for liver fibrosis therapy. Acta Biomater. 2022;152:235–54.
Zhang Z, Wang C, Zha Y, Hu W, Gao Z, Zang Y, Chen J, Zhang J, Dong L. Corona-directed nucleic acid delivery into hepatic stellate cells for liver fibrosis therapy. ACS Nano. 2015;9(3):2405–19.
Yang J, Hou Y, Ji G, Song Z, Liu Y, Dai G, Zhang Y, Chen J. Targeted delivery of the RGD-labeled biodegradable polymersomes loaded with the hydrophilic drug oxymatrine on cultured hepatic stellate cells and liver fibrosis in rats. Eur J Pharm Sci. 2014;52:180–90.
Chen Z, Jain A, Liu H, Zhao Z, Cheng K. Targeted drug delivery to hepatic stellate cells for the treatment of liver fibrosis. J Pharmacol Exp Ther. 2019;370(3):695–702.
Boland ML, Laker RC, Mather K, Nawrocki A, Oldham S, Boland BB, Lewis H, Conway J, Naylor J, Guionaud S, Feigh M, Veidal SS, Lantier L, McGuinness OP, Grimsby J, Rondinone CM, Jermutus L, Larsen MR, Trevaskis JL, Rhodes CJ. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab. 2020;2(5):413–31.
Yang B, Chen Y, Shi J. Reactive oxygen species (ROS)-based nanomedicine. Chem Rev. 2019;119(8):4881–985.
Hoffmann MH, Griffiths HR. The dual role of reactive oxygen species in autoimmune and inflammatory diseases: evidence from preclinical models. Free Radical Biol Med. 2018;125:62–71.
Volarevic V, Paunovic V, Markovic Z, Simovic Markovic B, Misirkic-Marjanovic M, Todorovic-Markovic B, Bojic S, Vucicevic L, Jovanovic S, Arsenijevic N, Holclajtner-Antunovic I, Milosavljevic M, Dramicanin M, Kravic-Stevovic T, Ciric D, Lukic ML, Trajkovic V. Large graphene quantum dots alleviate immune-mediated liver damage. ACS Nano. 2014;8(12):12098–109.
Pinna A, Cali E, Kerherve G, Galleri G, Maggini M, Innocenzi P, Malfatti L. Fulleropyrrolidine-functionalized ceria nanoparticles as a tethered dual nanosystem with improved antioxidant properties. Nanoscale Adv. 2020;2(6):2387–96.
Surendran SP, Thomas RG, Moon MJ, Park R, Lee JH, Jeong YY. A bilirubin-conjugated chitosan nanotheranostics system as a platform for reactive oxygen species stimuli-responsive hepatic fibrosis therapy. Acta Biomater. 2020;116:356–67.
Li Y, Liang Q, Zhou L, Cao Y, Yang J, Li J, Liu J, Bi J, Liu Y. An ROS-responsive artesunate prodrug nanosystem co-delivers dexamethasone for rheumatoid arthritis treatment through the HIF-1α/NF-κB cascade regulation of ROS scavenging and macrophage repolarization. Acta Biomater. 2022;152:406–24.
Deng Y, Wang Y, Jia F, Liu W, Zhou D, ** Q, Ji J. Tailoring supramolecular prodrug nanoassemblies for reactive nitrogen species-potentiated chemotherapy of liver cancer. ACS Nano. 2021;15(5):8663–75.
Foglia B, Novo E, Protopapa F, Maggiora M, Bocca C, Cannito S, Parola M. Hypoxia, hypoxia-inducible factors and liver fibrosis. Cells. 2021;10(7):1764.
Ma Q, Reiter RJ, Chen Y. Role of melatonin in controlling angiogenesis under physiological and pathological conditions. Angiogenesis. 2020;23(2):91–104.
Iwakiri Y, Kim MY. Nitric oxide in liver diseases. Trends Pharmacol Sci. 2015;36(8):524–36.
Linnenberger R, Hoppstädter J, Wrublewsky S, Ampofo E, Kiemer AK. Statins and bempedoic acid: different actions of cholesterol inhibitors on macrophage activation. Int J Mol Sci. 2021;22(22):12480.
Hide D, Gil M, Andrade F, Rafael D, Raurell I, Bravo M, Barberá A, Gracia-Sancho J, Vargas V, Augustin S, Genescà J, Schwartz S Jr, Martell M. Simvastatin-loaded polymeric micelles are more effective and less toxic than conventional statins in a pre-clinical model of advanced chronic liver disease. Nanomedicine. 2020;29: 102267.
Hong F, Chou H, Fiel MI, Friedman SL. Antifibrotic activity of sorafenib in experimental hepatic fibrosis: refinement of inhibitory targets, dosing, and window of efficacy in vivo. Dig Dis Sci. 2013;58(1):257–64.
Liu S, Han D, Xu C, Yang F, Li Y, Zhang K, Zhao X, Zhang J, Lu T, Lu S, Shi C, Zhang R, Yang AG, Zhao A, Qin W, Yang B, Wen W. Antibody-drug conjugates targeting CD248 inhibits liver fibrosis through specific killing on myofibroblasts. Mol Med. 2022;28(1):37.
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–22.
Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials. 2009;30(29):5409–16.
Radmanesh F, Sadeghi Abandansari H, Ghanian MH, Pahlavan S, Varzideh F, Yakhkeshi S, Alikhani M, Moradi S, Braun T, Baharvand H. Hydrogel-mediated delivery of microRNA-92a inhibitor polyplex nanoparticles induces localized angiogenesis. Angiogenesis. 2021;24(3):657–76.
Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021;78(4):1233–61.
Gong J, Li J, Dong H, Chen G, Qin X, Hu M, Yuan F, Fang K, Wang D, Jiang S, Zhao Y, Huang W, Huang Z, Lu F. Inhibitory effects of berberine on proinflammatory M1 macrophage polarization through interfering with the interaction between TLR4 and MyD88. BMC Complement Altern Med. 2019;19(1):314.
Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD, Herrmann J. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine. 2018;30:303–16.
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–40.
Wang L, Li Y, Wang X, Wang P, Essandoh K, Cui S, Huang W, Mu X, Liu Z, Wang Y, Peng T, Fan GC. GDF3 protects mice against sepsis-induced cardiac dysfunction and mortality by suppression of macrophage pro-inflammatory phenotype. Cells. 2020;9(1):120.
He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S. IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience. 2020;437:161–71.
Kaps L, Leber N, Klefenz A, Choteschovsky N, Zentel R, Nuhn L, Schuppan D. In vivo siRNA delivery to immunosuppressive liver macrophages by α-mannosyl-functionalized cationic nanohydrogel particles. Cells. 2020;9(8):1905.
Liu S, Yu J, Zhang Q, Lu H, Qiu X, Zhou D, Qi Y, Huang Y. Dual cross-linked HHA hydrogel supplies and regulates MΦ2 for synergistic improvement of immunocompromise and impaired angiogenesis to enhance diabetic chronic wound healing. Biomacromolecules. 2020;21(9):3795–806.
Ono K, Sumiya M, Yoshinobu N, Dode T, Katayama T, Ueda N, Nagahama K. Angiogenesis promotion by combined administration of dfo and vein endothelial cells using injectable biodegradable, nanocomposite hydrogel scaffolds. ACS Appl Bio Mater. 2022;5(2):471–82.
Shi P, Kim YH, Mousa M, Sanchez RR, Oreffo ROC, Dawson JI. Self-assembling nanoclay diffusion gels for bioactive osteogenic microenvironments. Adv Healthcare Mater. 2018;7(15): e1800331.
Kiaee G, Dimitrakakis N, Sharifzadeh S, Kim HJ, Avery RK, Moghaddam KM, Haghniaz R, Yalcintas EP, Barros NR, Karamikamkar S, Libanori A, Khademhosseini A, Khoshakhlagh P. Laponite-based nanomaterials for drug delivery. Adv Healthcare Mater. 2022;11(7): e2102054.
Page DJ, Clarkin CE, Mani R, Khan NA, Dawson JI, Evans ND. Injectable nanoclay gels for angiogenesis. Acta Biomater. 2019;100:378–87.
Fercana GR, Yerneni S, Billaud M, Hill JC, VanRyzin P, Richards TD, Sicari BM, Johnson SA, Badylak SF, Campbell PG, Gleason TG, Phillippi JA. Perivascular extracellular matrix hydrogels mimic native matrix microarchitecture and promote angiogenesis via basic fibroblast growth factor. Biomaterials. 2017;123:142–54.
Liu Y, Dong Y, Wu X, Wang X, Niu J. Identification of immune microenvironment changes and the expression of immune-related genes in liver cirrhosis. Front Immunol. 2022;13: 918445.
Butcher MJ, Zhu J. Recent advances in understanding the Th1/Th2 effector choice. Faculty Rev. 2021;10:30.
Li RE, Hogervorst TP, Achilli S, Bruijns SC, Arnoldus T, Vivès C, Wong CC, Thépaut M, Meeuwenoord NJ, van den Elst H, Overkleeft HS, van der Marel GA, Filippov DV, van Vliet SJ, Fieschi F, Codée JDC, van Kooyk Y. Systematic dual targeting of dendritic cell C-type lectin receptor DC-SIGN and TLR7 using a trifunctional mannosylated antigen. Front Chem. 2019;7:650.
Nakanishi K, Tsukimoto M, Tanuma S, Takeda K, Kojima S. Silica nanoparticles activate purinergic signaling via P2X7 receptor in dendritic cells, leading to production of pro-inflammatory cytokines. Toxicol In Vitro. 2016;35:202–11.
Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7.
Tsugita M, Morimoto N, Tashiro M, Kinoshita K, Nakayama M. SR-B1 is a silica receptor that mediates canonical inflammasome activation. Cell Rep. 2017;18(5):1298–311.
Feray A, Guillet É, Szely N, Hullo M, Legrand FX, Brun E, Rabilloud T, Pallardy M, Biola-Vidamment A. Synthetic amorphous silica nanoparticles promote human dendritic cell maturation and CD4+ T-lymphocyte activation. Toxicol Sci. 2021;185(1):105–16.
Tomić S, Ðokić J, Vasilijić S, Ogrinc N, Rudolf R, Pelicon P, Vučević D, Milosavljević P, Janković S, Anžel I, Rajković J, Rupnik MS, Friedrich B, Colić M. Size-dependent effects of gold nanoparticles uptake on maturation and antitumor functions of human dendritic cells in vitro. PLoS ONE. 2014;9(5): e96584.
Fytianos K, Rodriguez-Lorenzo L, Clift MJ, Blank F, Vanhecke D, von Garnier C, Petri-Fink A, Rothen-Rutishauser B. Uptake efficiency of surface modified gold nanoparticles does not correlate with functional changes and cytokine secretion in human dendritic cells in vitro. Nanomed Nanotechnol Biol Med. 2015;11(3):633–44.
Yang D, Zhao Y, Guo H, Li Y, Tewary P, **ng G, Hou W, Oppenheim JJ, Zhang N. [Gd@C(82)(OH)(22)](n) nanoparticles induce dendritic cell maturation and activate Th1 immune responses. ACS Nano. 2010;4(2):1178–86.
Barillet S, Fattal E, Mura S, Tsapis N, Pallardy M, Hillaireau H, Kerdine-Römer S. Immunotoxicity of poly (lactic-co-glycolic acid) nanoparticles: influence of surface properties on dendritic cell activation. Nanotoxicology. 2019;13(5):606–22.
Tkach AV, Yanamala N, Stanley S, Shurin MR, Shurin GV, Kisin ER, Murray AR, Pareso S, Khaliullin T, Kotchey GP, Castranova V, Mathur S, Fadeel B, Star A, Kagan VE, Shvedova AA. Graphene oxide, but not fullerenes, targets immunoproteasomes and suppresses antigen presentation by dendritic cells. Small. 2013;9(9–10):1686–90.
Blank F, Gerber P, Rothen-Rutishauser B, Sakulkhu U, Salaklang J, De Peyer K, Gehr P, Nicod LP, Hofmann H, Geiser T, Petri-Fink A, Von Garnier C. Biomedical nanoparticles modulate specific CD4+ T cell stimulation by inhibition of antigen processing in dendritic cells. Nanotoxicology. 2011;5(4):606–21.
Giovanni M, Yue J, Zhang L, **e J, Ong CN, Leong DT. Pro-inflammatory responses of RAW264.7 macrophages when treated with ultralow concentrations of silver, titanium dioxide, and zinc oxide nanoparticles. J Hazard Mater. 2015;297:146–52.
Tabbasam R, Khursid S, Ishaq Y, Malik A. In vivo evaluation of inorganic nanoparticle complexes against CCL4 induced hepatotoxicity. Curr Drug Deliv. 2021;18(8):1197–203.
Yen HJ, Hsu SH, Tsai CL. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5(13):1553–61.
Laskar A, Eilertsen J, Li W, Yuan XM. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochem Biophys Res Commun. 2013;441(4):737–42.
Su L, Zhang W, Wu X, Zhang Y, Chen X, Liu G, Chen G, Jiang M. Glycocalyx-mimicking nanoparticles for stimulation and polarization of macrophages via specific interactions. Small. 2015;11(33):4191–200.
Perisé-Barrios AJ, Gómez R, Corbí AL, de la Mata J, Domínguez-Soto A, Muñoz-Fernandez MA. Use of carbosilane dendrimer to switch macrophage polarization for the acquisition of antitumor functions. Nanoscale. 2015;7(9):3857–66.
Kwon D, Cha BG, Cho Y, Min J, Park EB, Kang SJ, Kim J. Extra-large pore mesoporous silica nanoparticles for directing in vivo M2 macrophage polarization by delivering IL-4. Nano Lett. 2017;17(5):2747–56.
Wang X, Chang CH, Jiang J, Liu X, Li J, Liu Q, Liao YP, Li L, Nel AE, **a T. Mechanistic differences in cell death responses to metal-based engineered nanomaterials in Kupffer cells and hepatocytes. Small. 2020;16(21):e2000528.
Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44(3):439–49.
Farajzadeh R, Zarghami N, Serati-Nouri H, Momeni-Javid Z, Farajzadeh T, Jalilzadeh-Tabrizi S, Sadeghi-Soureh S, Naseri N, Pilehvar-Soltanahmadi Y. Macrophage repolarization using CD44-targeting hyaluronic acid-polylactide nanoparticles containing curcumin. Artif Cells Nanomed Biotechnol. 2018;46(8):2013–21.
Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The Neutrophil. Immunity. 2021;54(7):1377–91.
Lin MH, Lin CF, Yang SC, Hung CF, Fang JY. The interplay between nanoparticles and neutrophils. J Biomed Nanotechnol. 2018;14(1):66–85.
Masoud R, Bizouarn T, Trepout S, Wien F, Baciou L, Marco S, Houée Levin C. Titanium dioxide nanoparticles increase superoxide anion production by acting on NADPH oxidase. PloS ONE. 2015;10(12):e0144829.
Liang X, Li X, Duan J, Chen Y, Wang X, Pang L, Kong D, Song B, Li C, Yang J. Nanoparticles with CD44 targeting and ROS triggering properties as effective in vivo antigen delivery system. Mol Pharm. 2018;15(2):508–18.
Liang X, Duan J, Li X, Zhu X, Chen Y, Wang X, Sun H, Kong D, Li C, Yang J. Improved vaccine-induced immune responses via a ROS-triggered nanoparticle-based antigen delivery system. Nanoscale. 2018;10(20):9489–503.
Couto D, Freitas M, Vilas-Boas V, Dias I, Porto G, Lopez-Quintela MA, Rivas J, Freitas P, Carvalho F, Fernandes E. Interaction of polyacrylic acid coated and non-coated iron oxide nanoparticles with human neutrophils. Toxicol Lett. 2014;225(1):57–65.
Hou M, Wu X, Zhao Z, Deng Q, Chen Y, Yin L. Endothelial cell-targeting, ROS-ultrasensitive drug/siRNA co-delivery nanocomplexes mitigate early-stage neutrophil recruitment for the anti-inflammatory treatment of myocardial ischemia reperfusion injury. Acta Biomater. 2022;143:344–55.
Babin K, Antoine F, Goncalves DM, Girard D. TiO2, CeO2 and ZnO nanoparticles and modulation of the degranulation process in human neutrophils. Toxicol Lett. 2013;221(1):57–63.
Lu N, Sui Y, Tian R, Peng YY. Adsorption of plasma proteins on single-walled carbon nanotubes reduced cytotoxicity and modulated neutrophil activation. Chem Res Toxicol. 2018;31(10):1061–8.
Bozzano F, Perrone C, Moretta L, De Maria A. NK cell precursors in human bone marrow in health and inflammation. Front Immunol. 2019;10:2045.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86(3):513–28.
Khan MA, Khan A. Role of NKT cells during viral infection and the development of NKT cell-based nanovaccines. Vaccines. 2021;9(9):949.
Vis B, Hewitt RE, Monie TP, Fairbairn C, Turner SD, Kinrade SD, Powell JJ. Ultrasmall silica nanoparticles directly ligate the T cell receptor complex. Proc Natl Acad Sci USA. 2020;117(1):285–91.
Kheirolomoom A, Kare AJ, Ingham ES, Paulmurugan R, Robinson ER, Baikoghli M, Inayathullah M, Seo JW, Wang J, Fite BZ, Wu B, Tumbale SK, Raie MN, Cheng RH, Nichols L, Borowsky AD, Ferrara KW. In situ T-cell transfection by anti-CD3-conjugated lipid nanoparticles leads to T-cell activation, migration, and phenotypic shift. Biomaterials. 2022;281: 121339.
He S, Simpson BK, Sun H, Ngadi MO, Ma Y, Huang T. Phaseolus vulgaris lectins: a systematic review of characteristics and health implications. Crit Rev Food Sci Nutr. 2018;58(1):70–83.
Zupke O, Distler E, Jürchott A, Paiphansiri U, Dass M, Thomas S, Hartwig UF, Theobald M, Landfester K, Mailänder V, Herr W. Nanoparticles and antigen-specific T-cell therapeutics: a comprehensive study on uptake and release. Nanomedicine. 2015;10(7):1063–76.
Canakci M, Singh K, Munkhbat O, Shanthalingam S, Mitra A, Gordon M, Osborne BA, Thayumanavan S. Targeting CD4(+) cells with anti-CD4 conjugated mertansine-loaded nanogels. Biomacromolecules. 2020;21(6):2473–81.
Tombácz I, Laczkó D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A, Papp TE, Alameh MG, Shuvaev V, Mui BL, Tam YK, Muzykantov V, Pardi N, Weissman D, Parhiz H. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol Ther. 2021;29(11):3293–304.
Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable Lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020;20(3):1578–89.
Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, Cheung JF, Mao T, Askari AS, Chen S, Fitzgerald B, Foster GG, Eisenbarth SC, Zhao H, Craft J, Joshi NS. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 2021;184(25):6101-6118.e13.
Talamini L, Picchetti P, Ferreira LM, Sitia G, Russo L, Violatto MB, Travaglini L, Fernandez Alarcon J, Righelli L, Bigini P, De Cola L. Organosilica cages target hepatic sinusoidal endothelial cells avoiding macrophage filtering. ACS Nano. 2021;15(6):9701–16.
Sato Y, Kinami Y, Hashiba K, Harashima H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J Control Release. 2020;322:217–26.
Bezbaruah R, Chavda VP, Nongrang L, Alom S, Deka K, Kalita T, Ali F, Bhattacharjee B, Vora L. Nanoparticle-based delivery systems for vaccines. Vaccines. 2022;10(11):1946.
Zhu M, Wang R, Nie G. Applications of nanomaterials as vaccine adjuvants. Hum Vaccin Immunother. 2014;10(9):2761–74.
Yu H, Ji M. Recent advances of organic near-infrared II fluorophores in optical properties and imaging functions. Mol Imag Biol. 2021;23(2):160–72.
Henna TK, Pramod K. Graphene quantum dots redefine nanobiomedicine. Mater Sci Eng C Mater Biol Appl. 2020;110: 110651.
Hunt NJ, Lockwood GP, Le Couteur FH, McCourt PAG, Singla N, Kang SWS, Burgess A, Kuncic Z, Le Couteur DG, Cogger VC. Rapid intestinal uptake and targeted delivery to the liver endothelium using orally administered silver sulfide quantum dots. ACS Nano. 2020;14(2):1492–507.
Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30(6):507–19.
Ricciardi S, Manfrini N, Alfieri R, Calamita P, Crosti MC, Gallo S, Müller R, Pagani M, Abrignani S, Biffo S. The translational machinery of human CD4(+) T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab. 2018;28(6):895-906.e5.
Buszko M, Shevach EM. Control of regulatory T cell homeostasis. Curr Opin Immunol. 2020;67:18–26.
Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454.
Tan S, Li S, Min Y, Gisterå A, Moruzzi N, Zhang J, Sun Y, Andersson J, Malmström RE, Wang M, Berggren PO, Schlisio S, Liao W, Ketelhuth DFJ, Ma C, Li N. Platelet factor 4 enhances CD4(+) T effector memory cell responses via Akt-PGC1α-TFAM signaling-mediated mitochondrial biogenesis. J Thrombosis Haemostasis. 2020;18(10):2685–700.
Slattery K, Gardiner CM. NK Cell Metabolism and TGFβ—implications for immunotherapy. Front Immunol. 2019;10:2915.
Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol. 2019;10:2099.
Qing J, Zhang Z, Novák P, Zhao G, Yin K. Mitochondrial metabolism in regulating macrophage polarization: an emerging regulator of metabolic inflammatory diseases. Acta Biochim Biophys Sin. 2020;52(9):917–26.
Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, Ying T, Fixman ED, Jones RG, McCaffrey LM, Krawczyk CM. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nature Commun. 2018;9(1):2463.
**a L, Oyang L, Lin J, Tan S, Han Y, Wu N, Yi P, Tang L, Pan Q, Rao S, Liang J, Tang Y, Su M, Luo X, Yang Y, Shi Y, Wang H, Zhou Y, Liao Q. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20(1):28.
Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience. 2018;5:99–109.
Shevyrev D, Tereshchenko V. Treg heterogeneity function, and homeostasis. Front Immunology. 2019;10:3100.
Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–43.
Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302(1):259–72.
Acknowledgements
Not application
Funding
This study was supported by the National Natural Science Foundation of China (NO. 8189101, 81891012, 81630101 and U19A2010, China), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (NO. ZYYCXTD-D-202209, China), Sichuan TCM Science and Technology Industry Innovation Team (NO. 2022C001), and Sichuan Science and Technology Program (NO. 2021JDRC0041, 22ZYZYTS0071 China).
Author information
Authors and Affiliations
Contributions
XZ and FKA collected relevant literature, drafted manuscripts; XZ and XX prepared figures; CW, ZC, SD reviewed the manuscript; YL and CP guided the preparation of this manuscript and provides funding support. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The first affiliation has been revised.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, X., Amevor, F.K., Xue, X. et al. Remodeling the hepatic fibrotic microenvironment with emerging nanotherapeutics: a comprehensive review. J Nanobiotechnol 21, 121 (2023). https://doi.org/10.1186/s12951-023-01876-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-023-01876-5