
Abstract
Cancer drug resistance represents the main obstacle in cancer treatment. Drug-resistant cancers exhibit complex molecular mechanisms to hit back therapy under pharmacological pressure. As a reversible epigenetic modification, N6-methyladenosine (m6A) RNA modification was regarded to be the most common epigenetic RNA modification. RNA methyltransferases (writers), demethylases (erasers), and m6A-binding proteins (readers) are frequently disordered in several tumors, thus regulating the expression of oncoproteins, enhancing tumorigenesis, cancer proliferation, development, and metastasis. The review elucidated the underlying role of m6A in therapy resistance. Alteration of the m6A modification affected drug efficacy by restructuring multidrug efflux transporters, drug-metabolizing enzymes, and anticancer drug targets. Furthermore, the variation resulted in resistance by regulating DNA damage repair, downstream adaptive response (apoptosis, autophagy, and oncogenic bypass signaling), cell stemness, tumor immune microenvironment, and exosomal non-coding RNA. It is highlighted that several small molecules targeting m6A regulators have shown significant potential for overcoming drug resistance in different cancer categories. Further inhibitors and activators of RNA m6A-modified proteins are expected to provide novel anticancer drugs, delivering the therapeutic potential for addressing the challenge of resistance in clinical resistance.
Similar content being viewed by others


Introduction
Estimated 600,000 people die from cancer each year, which is still a challenging problem that scientists are desperate to resolve [1, 2]. Oncotherapy is currently divided into five mainstream approaches: surgical resection, chemotherapy, radiotherapy, biological immunotherapy, and targeted therapy [3, 4]. Although there have been numerous breakthroughs for specific cancer categories, most strategies still are not as effective as expected. The major reason for treating cancer failure is the lacked understanding of the molecular mechanisms of therapeutic resistance. Resistance to chemotherapy drugs is usually divided into two main categories: acquired and intrinsic [5]. Intrinsic resistance, also called primary resistance, is a consequence of genetic alterations before treatment. Acquired drug resistance is caused by drug treatment and is also known as secondary resistance. Both are due to mutations and/or epigenetic changes in the genome of cancer cells. In the process of drugs binding to target and function, multiple mechanisms must be involved, including altered metabolism, transport, and varied target proteins [6]. Additionally, impaired apoptosis, augmented populations of cancer stem cells (CSCs), altered expression of oncogene/tumor suppressors, and manipulated tumor immune microenvironment (TIME) are also the dominant causes in charge of diminishing antitumor drug efficacy [7, 8]. Nevertheless, these are only influencing factors of therapy-resistant cancers, and the specific mechanism for therapy-resistant are unknown.
Researchers have identified more than 160 different chemically RNA modifications, creating a novel frontier called epitranscriptomics [9]. N6-methyladenosine (m6A) RNA modification has been identified as one of the most pervasive and abundant RNA modifications in eukaryotic messenger RNA (mRNA) [10, 11] and viral nuclear RNA [12, 13] since discovered in the 1970s. The process of m6A modification is dynamic and reversible, which is regulated by methylases (“writers”) and demethylases (“erasers”) (Table 1). m6A is installed by writers including methyltransferase-like (METTL) 3 [14], METTL14 [15], Wilms tumor 1-associated protein (WTAP) [17], KIAA1429 [18], METTL16 [16], RBM15 [20], and ZC3H13 [21]. m6A is removed by erasers such as fat mass and obesity-associated protein (FTO) [22] and alkB homolog 5 (ALKBH5) [23]. Different families of m6A reader proteins are capable of recognizing RNAs modified with m6A. One type of natural m6A reader protein contains the YT521-B homology (YTH) domain [33], and heterogeneous nuclear ribonucleoproteins (HNRNPs) belong to the other type, which mainly regulated alternative splicing or processing of target transcripts [29]. Other subfamily members are insulin-like growth factor 2 (IGF2) mRNA binding proteins (IGF2BP1/2/3) [31], and eIF3 [32].
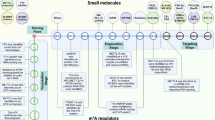
Emerging evidence indicated that m6A modifications were strongly associated with therapy resistance. In several neoplasms, m6A regulators (writers, erasers, and readers) are frequently overexpressed, regulating oncoprotein expression, enhancing cancer inception, and cell multiplication [34]. m6A modulates multiple anticancer resistance, including drug transport and metabolism, target receptors, cancer stemness, DNA damage repair, and cell death [35,36,37,38]. In addition, m6A is closely related to the immune response in the tumor microenvironment, providing new prospects for tumor immunotherapy [39]. Importantly, small-molecule activators and inhibitors of m6A regulators have recently been revealed to possess considerable anticancer effects when applied alone or in combination with other anticancer agents, suggesting the novel function of m6A in anticancer drug resistance [40]. This review primarily introduced the significant role of m6A modification in tumor drug resistance, reviewed the mechanisms of RNA m6A modification associated with drug resistance, and further discussed the strategies targeting the m6A change in predicting and treating cancer resistance (Fig. 1).

m6A-mediated biological processes of drug resistance. m6A was involved in several aspects of drug pharmacokinetics. m6A modifications upregulated drug transporters (e.g., ABCB1, ABCC1, ABCC10), facilitating ATP-driven drug efflux. m6A was also engaged in regulating several drug-metabolizing enzymes (e.g., CYP2C8 and UGT2B7) that affected the efficacy of chemotherapeutic drugs. Some drug targets (e.g., EGFR) were regulated by m6A and affected cancer development. Additionally, m6A also participated in activating downstream effects, which were embodied in the following three aspects. Firstly, m6A could selectively upregulate the p53 (R273 H) protein, releasing prohibited anti-apoptotic proteins (e.g., BCL-2, IAPs). Secondly, m6A altered the expression of various key signaling molecules (e.g., ULK1, FOXO3) in autophagy and ultimately regulated autophagy through light chain 3-II (LC3-II). Thirdly, m6A modification activated oncogenic bypass signaling through key molecules (e.g., IGF1R, DUXAP8) and promoted cell stemness, which became an important barrier to drug resistance. Immune cell infiltration and cytokine secretion in the tumor microenvironment were also regulated by m6A, which was relevant for cancer immunotherapy. The m6A modification of exosomal non-coding RNA was implicated in multiple biological processes in tumors and was associated with resistance to multiple anticancer drugs
Mechanisms of m6A-mediated drug resistance
Cancer resistance is caused by a variety of factors, such as individual differences in drug sensitivity, tumor location, tissue spectrum, tumor aggressiveness, and alterations in intracellular molecules [3, 41]. The mechanism of m6A-mediated drug resistance was embodied in drug pharmacokinetics, tumor cells, and tumor microenvironment. Deciphering the impact of m6A modifications on the mechanisms of resistance to anticancer therapy could offer more prospects for individualized tumor treatment.
m6A modulation in drug pharmacokinetics
m6A modulated aberrant drug transport and metabolism
Several membrane transporter proteins work together to promote drug efflux and resistance to chemotherapeutics. Most drug efflux experiments have focused on the role of the ATP-binding cassette (ABC) proteins [42]. Multidrug resistance (MDR) is mediated by a wide range of ABC transporters, such as ABCB1 (MDR1), ABCC1 (MRP1), ABCC10 (MRP7), and others [43, 44]. Recently, researchers have demonstrated that RNA m6A modifications regulated the expression of ABC family proteins through either direct impact on tumor transcripts or indirect effects on upstream signaling pathways. For instance, m6A upregulated estrogen-related receptor gamma (ERRγ) in chemo-resistant cancer cells. ERRγ not only directly enhanced ABCB1 transcription but also indirectly by further strengthening the interaction with p65 [45]. Besides, METTL3 m6A-dependently enhanced translation of ABCD1, leading to migration and spheroid formation in clear cell renal cell carcinoma (ccRCC) [46]. Notably, exosomal-FTO facilitated ABCC10 of recipient cells via FTO/YTHDF2/ABCC10 axis, eventually leading to gefitinib resistance in non–small cell lung cancer (NSCLC) [47]. Excluding drug transport, the efficacy of chemotherapeutic drugs is determined by the effects of drug metabolism, such as bioactivation, catabolism, conjugation, and elimination [48]. Recent studies have revealed that the m6A modification had a negative regulatory effect on regulating drug metabolism. For example, METTL3/14 depletion upregulated cytochrome P450 family member cytochrome P450 2C8 (CYP2C8), whereas FTO depletion suppressed it. Mechanically, YTHDC2 promoted CYP2C8 mRNA degradation by recognizing the m6A in CYP2C8 mRNA [49]. Another drug metabolism enzyme, carboxylesterase 2 (CES2), exhibits the exact mechanism of negative regulation by m6A as CYP2C8 [50]. UDP-glucuronosyltransferases (UGTs) are enzymes that catalyze the glucuronidation of various endogenous and exogenous compounds. In Huh-7 cells, the m6A regulator-mediated methylation modification also showed a negative correlation with UGT2B7 [51]. In summary, m6A modifications are novel regulators of drug transport and metabolism, contributing to the practice of personalized medicine.
m6A drove drug target alterations
Alterations to drug targets, such as mutations or changes in expression levels, impact drug response and resistance [52]. For example, the TP53 gene coding for the p53 protein and mutant p53 proteins augmented cancer progression and generated drug resistance. METTL3-mediated m6A produced the p53 R273H mutant protein, causing MDR in colon cancer cells (Fig. 1) [53]. Epidermal growth factor receptor (EGFR) is another potential therapeutic target whose activation led to tumor cell proliferation, evasion of apoptosis, angiogenesis, and metastasis [54]. METTL3 augmented the translation efficiency of EGFR, followed by rebound activation of RAF/MEK/ERK, resulting in acquired PLX4032 resistance in melanoma (Fig. 1) [55]. Furthermore, YTHDF1 and YTHDF2 impacted cancer via binding m6A sites in the 3′-UTR of EGFR transcription and contributed to aberrant activities of downstream signal pathways [56, 57]. m6A-induced alterations in p53 protein and EGFR drug targets affect the efficacy of anticancer drugs, which may enable us to develop effective strategies to reverse the alterations in drug targets.
m6A modulation in tumor cells
m6A regulated DNA damage repair
An ocean of chemotherapeutic agents primarily targeting genomic DNA can result in DNA lesions and inhibit transcription and replication [58]. m6A methyltransferase METTL3 facilitated oxaliplatin resistance in gastric cancer (GC) stem cells by substantial DNA damage repair [59]. Furthermore, METTL3 enhanced the expression of UBE2B, a crucial enzyme involved in DNA damage repair, thereby triggering multifarious drug resistance [60,61,62]. Additionally, other m6A regulators, YTHDF1 and ALKBH5, were also engaged in chemoresistance (including adriamycin, cisplatin, and olaparib) by enhancing DNA damage repair in breast cancer (BC) [63, 64].
m6A activated downstream effects
Anticancer drugs result in tumor cells’ death upon binding to their cellular targets. The m6A modification affected a diverse array of downstream impacts, including demolition of apoptosis, activation of autophagy, and energizing of oncogenic bypass signaling, which was a crucial part of current cancer therapy [65, 66].
m6A mediated cell apoptosis
Cell sensitivity to anticancer drugs was primarily determined by the upregulation of anti-apoptotic proteins, including B-cell lymphoma 2 (BCL-2), IAPs, and FLIP [67, 68]. Remarkably, m6A modification had a differential effect on BCL-2 expression according to the type of cancer. Recent research revealed that overexpression of FTO was accompanied by BCL-2 upregulation [69], which was consistent with the trend of regulation of BCL-2 by ALKBH5 found in epithelial ovarian cancer (EOC) [70]. Consequently, RNA m6A modification was inversely correlated with BCL-2 expression and anti-apoptosis. Nonetheless, varied results were found that m6A also positively influenced the expression of anti-apoptotic proteins. Wang et al. found METTL3 knockdown dramatically augmented apoptosis capabilities in BC by decreasing BCL-2 expression [71]. In esophageal cancer, NSCLC, and GC, reduced expression of m6A positively correlated with the decrease of the anti-apoptotic protein BCL-2, contributing to the activation of apoptosis [72,73,74]. Overall, the m6A modification modulated apoptosis based on the cancer context, uncovering the dual role of m6A in tumor cells.
m6A mediated cell autophagy
Autophagy is a lysogenic process that permits cells to own stress-co** strategies by degrading damaged organelles and accumulated proteins, which could result in cancer resistance treated with anticancer drugs [75,76,77,78]. m6A modification acted as a double-edged sword in autophagy regulation. In some cases, the RNA m6A modification inhibited autophagy (Fig. 2A). Light chain 3B (LC3B) was a well-known autophagy biomarker in the cytoplasmic matrix [79]. In hepatocellular carcinoma (HCC), METTL3 depletion promoted the LC3-II accumulation by reducing the stability of FOXO3 mRNA through a YTHDF1-dependent mechanism [80]. ** et al. [81] validated that FTO enhanced LC3B II accumulation by slowing the decay rate of unc-51-like kinase 1 (ULK1) transcripts in a YTHDF2-dependent manner. By the same mechanism, FTO enhanced the translation of autophagy-associated gene-5 (ATG5) and ATG7 mRNAs and promoted an increase of LC3-II [82]. Conversely, m6A modification promoted autophagy in some cases (Fig. 2B). ALKBH5 activated the EGFR-PIK3CA-AKT-mTOR pathway and specifically cemented the BCL-2 mRNA stability to slow the autophagy in EOC [70]. The latest study found that m6A reader YTHDF3 promotes autophagy by recognizing the METTL3-mediated m6A modification site around the FOXO3 mRNA stop codon, providing new evidence for a dual role in m6A autophagy [83].

Dual effects of m6A in autophagy. On the one hand, the m6A modification inhibits autophagy. In hepatocellular carcinoma (HCC), METTL3 enhanced forkhead box O3 (FOXO3) mRNA stability and inhibited light chain 3-II (LC3-II) accumulation through a YTHDF1-dependent mechanism. The overexpression of FTO induced YTHDF2-dependent inhibition of unc-51-like kinase 1 (ULK1) mRNA decay and promoted LC3-II accumulation and autophagy. With the help of YTHDF2, FTO also increased the translation of autophagy-associated gene-5 (ATG5) and ATG7 mRNAs and promoted autophagosome assembly. On the other hand, m6A modification also promotes autophagy. In epithelial ovarian cancer (EOC), ALKBH5 slowed autophagy by cementing B-cell lymphoma 2 (BCL-2) mRNA stability and activating the EGFR-PIK3CA-AKT-mTOR pathway. Additionally, the m6A reader YTHDF3 promoted autophagy through the upregulation of FOXO3 mRNA translation
m6A regulated oncogenic bypass signaling
Even though targeted therapies enabled tumor cells to be sensitive to chemotherapy, drug resistance remained a significant obstacle owing to the activation of oncogenic bypass pathways (including Wnt/β-catenin, PI3K/AKT, MAPK, or c-MET signaling) [84,85,86]. ALKBH5 suppressed m6A modification of the WIF-1 mRNA to promote its transcription, which probably interfered with the Wnt signaling and led to chemosensitivity [87]. Besides, Xu et al. [88] revealed that the elevated level of m6A in circular RNA (circRNA)-SORE enhanced its stability, allowing it to induce sorafenib resistance by acting as a microRNA (miRNA) sponge to isolate miR-103a-2-5p and miR-660-3p, thereby competitively activating the Wnt/β-catenin pathway. YTHDC2, the m6A reader protein, regulated irradiation efficacy via IGF1R-AKT/S6 pathway, leading to radiotherapy resistance of nasopharyngeal carcinoma (Fig. 1) [89]. Alternatively, m6A modification-mediated DUXAP8 regulated malignant phenotype and chemoresistance of HCC through miR-584-5p/MAPK1/ERK pathway (Fig. 1) [90]. Beyond that, chidamide reduced c-MET expression by lowering m6A methylation, which increased crizotinib sensitivity in NSCLC cells in a c-MET/HGF-dependent manner [91]. NF-κB activating protein (NKAP), as a reader of m6A, promoted SLC7A11 mRNA splicing and maturation, thereby enhancing cell resistance to ferroptosis inducers [92]. Overall, the m6A mutation activated the oncogenic bypass pathway, circumventing the classical drug targets, which could be considered in targeted therapy to avoid or overcome drug resistance (Fig. 3).

m6A-regulated oncogenic bypass signaling. Downregulation of ALKBH5 led to the downregulation of WIF-1 mRNA expression, thus activating the Wnt pathway. The elevated levels of m6A in circRNA-SORE enhanced its stability and allowed it to competitively activate the Wnt/β-certain pathway by acting as a miRNA sponge. YTHDC2 promoted radiotherapy resistance by activating the IGF1R-AKT/S6 signaling axis. m6A modification-mediated DUXAP8 contributed to chemoresistance via miR-584-5p/MAPK1/ERK. Chidamide decreased c-MET expression and increased crizotinib sensitivity by reducing m6A methylation. NKAP promoted SLC7A11 mRNA splicing and maturation, thereby inhibiting ferroptosis
m6A affected the sustainment of cell stemness
CSCs represent a small population of tumor cells sustaining versatility and promoting tumor progression and drug resistance [93, 94]. METTL3 was involved in regulating the stemness and chemosensitivity of colon cancer through the upregulation of LGR5 [95]. Aside from that, METTL3 facilitated oxaliplatin resistance in CD133+ stem cells by promoting PARP1 mRNA stability and increased base resection repair pathway activity [59]. Liu and his team [131].
Targeting demethylase
FTO
Demethylase FTO played an oncogenic role in BC, AML, and other malignant tumors [149,150,151]. FTO-mediated m6A modification was also associated with drug resistance in various cancers, such as MM, glioblastoma, and melanoma. YAN et al. [69] confirmed that the TKI-tolerance phenotype emerged in leukemia patients because the overexpression of FTO caused m6A reduction. Signal transducers and activators of transcription 3 (STAT3) were constitutively active in several cancer types, and such hyperactivity was associated with an adverse clinical outcome [152]. Wang et al. [134] found increased expression of FTO and STAT3 in doxorubicin-resistant BC cells, and STAT3 bound to the FTO promoter to positively accommodate FTO expression. Moreover, FTO was involved in STAT3-mediated doxorubicin resistance and impaired doxorubicin sensitivity in BC cells. The overexpressing of FTO in cervical squamous cell carcinoma (CSCC) was resistant to radiotherapy and chemotherapy by the FTO-mediated mRNA demethylation and ERCC1 activity [135]. Interestingly, FTO was set up at high concentrations in patients’ MM cells and bone marrow tissues. Further analysis showed that FTO promoted bortezomib resistance by destabilizing SOD2 expression through an m6A-dependent manner, which might open up innovative therapeutic options [133]. JPX, a non-coding RNA adjacent to the X-inactive specific transcript, was entangled in tumor progression. It appeared that JPX interacted with the mRNA of phosphoinositide-dependent kinase-1 (PDK1) and promoted its stability and expression. Furthermore, JPX demethylated PDK1 mRNA, through its interaction with FTO alpha-ketoglutarate-dependent dioxygenase, contributed to the enhanced demethylation. Consequently, JPX exerted its GBM positive effects via the FTO/PDK1 axis and directly stabilized the PDK1 mRNA in temozolomide drug resistance [132]. Besides, the knockdown of FTO decreased the stability of PD-1, CXCR4, and SOX10, increasing RNA attenuation via m6A reader YTHDF2. It also sensitized melanoma cells to IFN-γ and anti-PD-1 therapy.
ALKBH5
ALKBH5, another m6A modification demethylase, was related to the onset, development, and prognosis of colon cancer, BLCA, EOC, and oral squamous cell carcinoma (OSCC) [153,154,155]. The downregulation of FTO and ALKBH5 in ovarian cancers with breast-cancer susceptibility gene 2 (BRCA2) mutations enhanced FZD10 mRNA m6A modifications, which ultimately reduced the sensitivity of PARPi via the Wnt/β-catenin pathway [138]. Moreover, ALKBH5 promoted cisplatin resistance in cancer cells [136]. HOXA10, the upstream transcription factor of ALKBH5, could form a loop with ALKBH5. In this way, ALKBH5 and HOXA10 together activated the JAK2/STAT3 signaling pathway, mediating JAK2 m6A demethylation and promoting EOC resistance to cisplatin. A recent study found that ubiquitin-specific proteases (USPs) were associated with T-cell acute lymphoblastic leukemia (T-ALL) occurrence and chemoresistance. ALKBH5 exhibited a carcinogenic effect on cancers and improved USP mRNA’s stability, resulting in GC resistance [137]. Multiple neoplasms expressed the human RNA helicase DDX3, essential for cell proliferation, invasion, and metastasis. By directly regulating ALKBH5, DDX3 could decrease m6A methylation of FOXM1 and NANOG transcripts, giving rise to cisplatin resistance in OSCC cells [139]. Likewise, the deletion of the m6A demethylase ALKBH5 sensitized tumors to cancer immunotherapy, suggesting that ALKBH5 may be a potential target to improve the outcome of immunotherapy for melanomas, CRC, and other underlying cancers [106]. In pancreatic cancer (PC), ALKBH5-mediated m6A modification caused DDIT4-AS1 overexpression, and DDIT-AS1 increased cancer stemness and led to gemcitabine resistance by destabilizing DDIT4 and activating the mTOR pathway [156].
Targeting other m6A regulators
So far, strategies targeting m6A mainly relied on the regulation of methyltransferase (such as METTL3 and WTAP) and demethylase. However, multiple sources of evidence suggested that other m6A modulators also had great potential as drug-therapeutic targets. For instance, the depletion of METTL14, core subunits of RNA methyltransferase, dramatically slowed tumor growth and prolonged the survival in mice bearing CT26 CRC and B16 melanoma [101]. m6A reader protein also played a pivotal role in drug resistance. In NSCLC, Keap1 was degraded following YTHDF1 depletion, facilitating Keap1-Nrf2-AKR1C1 axis cells and resulting in cisplatin resistance [140]. MicroRNA-145 could abrogate YTHDF2’s role as an oncogene in HepG2 cells associated with HCC [157]. In CRC, hypoxia-induced antisense lncRNA STEAP3-AS1 competed with YTHDF2 to STEAP3 mRNA binding site, protecting STEAP3 mRNA from m6A-mediated degradation and leading to high STEAP3 protein expression. Followed by this, activation of the Wnt/β-catenin pathway contributed to CRC progression [158]. Moreover, paclitaxel, 5-FU, and cisplatin were more effective in cell lines that lacked the m6A reader protein HNRNPC [30]. IGF2BP3, another m6A reader, was bound to the m6A modification region of ABCB1 mRNA and increased chemoresistance in CRC cells [141]. These studies illustrated that HNRNPC and IGF2BP3 could be latent biomarkers for chemoresistance.
m6A-targeted compounds
FTO inhibitors
Rhein was the first identified inhibitor for FTO in vitro and in vivo, which was neither a structural mimic of 2OG nor a chelator of the metal ion. Rhein blocked FTO demethylase by competitively binding its catalytic domain instead [159]. In therapy, the rhein-TKI combination synthetically eradicated relapsed/refractory leukemia [69], while rhein exposure increased the level of m6A in leukemia. In contrast, no growth arrest was observed after 24 hours of 20 μM rhein, proposing the anticancer therapy of rhein. Ascorbic acid also enhanced the activity of 2OG-dependent dioxygenases. In BC, ascorbic acid analog MO-I-500 exhibited antiproliferative activity in an FTO-dependent manner [160, 161]. However, rhein, as well as MO-I-500, was a broad-spectrum 2-OG inhibitor, which tremendously reduced their applications. In a high-throughput fluorescence polarization assay, meclofenamic acid (MA), a non-steroidal anti-inflammatory drug, was selected as the inhibitor of FTO. Moreover, the ethyl ester form of MA (MA2) upgraded levels of m6A modification in mRNA [162]. Additionally, MA2 inhibited self-renewal and tumorigenesis of GSCs in a GSC-xenograft mouse model and prolonged survival [163]. Of note, MA2 enhanced the antitumor effect of chemotherapy in glioma [164]. As a result of the specific inhibitory property of MA, higher potency derivatives were designed and synthesized. A new MA-derived inhibitor, FB23, directly bound to FTO and selectively inhibited its activity, which possessed 140-fold over that of MA. The benzohy-droxamic acid, termed FB23–2, was a further practical analog of FB23 [165]. FB23–2 exhibited FTO-dependent anti-leukemia effects broadly and targeted the same signaling pathways as FB23. Dac51, another small-molecule analog of FB23, could modulate the tumor microenvironment via inhibiting FTO and mounting CD8+ T cell infiltration, contributing to a remarkable antitumor efficac y[105]. FTO-04 demonstrated robust inhibition of neurosphere formation in patient-derived GSCs but did not inhibit the growth of healthy human neural stem cells. On the side, FTO-04-mediated inhibition of FTO increased m6A modification and demethylated N6,2′-O-dimethyladenosine (m6Am) levels of GSCs [166]. Nafamostat mesylate often was applied in treating pancreatitis and cancers. The combination of thermodynamic and enzymatic activity provided insight into the FTO inhibition of nafamostat mesylate [167]. R-2-hydroxyglutarate (R-2HG) was architecturally and chemically similar to another inhibitor, 2OG. R-2HG inhibited FTO’s enzymatic activity by competitive inhibition and proved the overall antitumor effect. As a result of the R-2HG therapeutic regimen, m6A modification levels increased. Meanwhile, aerobic glycolysis was suppressed by inhibiting FTO activity and downstream signaling molecules, consisting of MYC, CEBPA, PFKP, and LDHB [168, 169]. CS1 and CS2 displayed a much higher efficacy. Consequently, two highly efficacious FTO inhibitors were named CS1 and CS2. They displayed a much higher efficacy in inhibiting AML cells’ viability than two previously reported FTO inhibitors (FB23–2 and MO-I-500) [103]. Therefore, FTO represented a modern therapeutic potential to target cancer therapy, and more clinical studies were required to confirm the long-term side effects of these inhibitors.
METTL3 inhibitors
Bedi et al. [170] reported a virtual screening method for almost 4000 adenosine derivatives to identify potential METTL3 inhibitors. Their best compound, S-adenosyl-L-methionine (SAM) mimic, was the first small molecule to inhibit METTL3. METTL3 inhibitors possessed excellent ligand efficiency, and their binding patterns were validated by protein crystallography. Respective RNA m6A methyltransferase inhibitors displayed anticancer abilities. Accompanied by the selective reduction of m6A levels on known leukemogenic mRNAs, STM2457 treatment reduced AML growth and increased differentiation and apoptosis [171]. Another METTL3 chemical inhibition, UZH1a, reduced the m6A/A ratio in mRNAs of different cell lines, revealing the potential implications of METTL3 inhibition in tremendous disease models [172].
Other m6A regulator activators and inhibitors
Using silico-based discovery could identify small-molecule ligands binding to the METTL3–14-WTAP complex. Primarily, SAM bonded with Asp377 and acted as a hydrogen bond donor to the Asp395 of METTL3 protein. Similarly, four compounds bound to the extent of the METTL3 enzyme relating to Asp295, Phe534, Arg536, and Asn539. METTL3-METTL14 RNA m6A methyltransferase complex activators provoked cells to modify mRNA m6A [173]. Their potential anticancer effects needed more experiments to prove. Li and his team [106] identified a small molecule inhibitor of ALKBH5 by using the X-ray crystal structure in silico screening of compounds and named ALK-04. Compound libraries verified this specific inhibitor. Subsequent proof found that melanoma tumor growth was significantly reduced in mice applying the ALK-04 compared to the control group. This study also provided evidence for ALKBH5 inhibitors combined with immunotherapy against melanoma. BTYNB has been identified by compound library screening with its ability to inhibit c-Myc and IGF2BP1 protein selectively [174]. The small molecule BTYNB also destabilized E2F1 mRNAs by impairing the IGF2BP1-RNA association, which interfered with cellular protein synthesis and tumor growth [174]. Table 3 collates the identified m6A-targeted compounds.
Conclusion and perspective
Despite considerable research underway to understand the function of m6A modifications in cancer proliferation and drug resistance, many questions remain unanswered. For example, as a broad RNA modification in eukaryotic messenger RNA, will the m6A regulator targeted compounds be a good candidate in tumor therapy? How to focus and target key molecules? How to specifically target the regulatory axis involved in m6A to reverse drug resistance in tumor tissue?
The practical significance of m6A modifications and regulators heralded a new dawn for targeting m6A regulators in therapy. However, few m6A-phenotype associated inhibitors and activators are clinically applicable. Followings might be responsible for this plight. Firstly, due to lacking study on cellular activity, how these compounds actually affect methylation levels is elusive. Secondly, adenosine analogs have poor cell permeability and pharmacokinetics, complicating their potential use. Thirdly, tumor heterogeneity and rare predictors mound a barrier between the targeted compounds and distinct cancers, contributing to poor clinical applicability. Therefore, further screening of potential agents is needed. For the precise regulation of m6A modifications (global and/or targeted), protein-protein interactions (PPI) or protein-nucleotide interactions would be promising strategies. Further studies on tumor biology, the development of high-quality chemical probes, and preclinical studies will help to identify precise biomarkers, which are crucial for individualized treatment, improved outcomes, and potential toxicity prediction. In addition, most of the reported targeted compounds are cytotoxic, whereas non-cytotoxic inhibitors that modulate the immune system also represent a promising combination. For example, the ALKBH5 inhibitor ALK-04 showed significant synergy with anti-PD-1 therapy while without cytotoxicity in vivo. Overall, the clinical application of compounds targeting m6A is still in its infancy. As the understanding of epigenomics in cancer grows, there is great promise for those therapy-resistant patients accompanied with abnormal m6A manners.
Availability of data and materials
Not applicable.
Abbreviations
- 5-FU:
-
5-fluorouracil
- ABC:
-
ATP-binding cassette
- ADR:
-
Adriamycin resistance
- ALKBH5:
-
alkb homolog 5
- AML:
-
Acute myeloid leukemia
- ATG:
-
Associated gene
- BC:
-
Breast cancer
- BCL-2:
-
B-cell lymphoma 2
- BLCA:
-
Bladder cancer
- BRCA2:
-
Breast-cancer susceptibility gene 2
- CAFs:
-
Cancer-associated fibroblasts
- ccRCC:
-
Clear cell renal cell carcinoma
- CES2:
-
Carboxylesterase 2
- circRNA:
-
Circular RNA
- CRC:
-
Colorectal cancer
- CSCC:
-
Cervical squamous cell carcinoma
- CSCs:
-
Cancer stem cells
- CYP2C8:
-
Cytochrome P450 2C8
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- EOC:
-
Epithelial ovarian cancer
- ERRγ:
-
Estrogen-related receptor gamma
- ESCC:
-
Esophageal squamous cell carcinoma
- FTO:
-
Fat mass and obesity-associated protein
- GBM:
-
Glioblastoma multiforme
- GC:
-
Gastric cancer
- GSCs:
-
Glioblastoma stem cells
- HCC:
-
Hepatocellular carcinoma
- HNRNP:
-
Heterogeneous nuclear ribonucleoprotein
- ICC:
-
Intrahepatic cholangiocarcinoma
- IGF2:
-
Insulin-like growth factor 2
- IFN-γ:
-
Interferon-gamma
- LC3B:
-
Light chain 3B
- lncRNA:
-
Long-noncoding RNA
- LUAD:
-
Lung adenocarcinoma
- m6A:
-
N6-methyladenosine
- m6Am :
-
Demethylate N6,2′-O-dimethyladenosine
- MA:
-
Meclofenamic acid
- MDR:
-
Multidrug resistance
- METTL:
-
Methyltransferase-like
- miRNA:
-
microRNA
- MM:
-
Multiple myeloma
- mRNA:
-
Messenger RNA
- NKAP:
-
NF-κB activating protein
- NKTCL:
-
Natural killer/T-cell lymphoma
- NSCLC:
-
Non–small cell lung cancer
- OSCC:
-
Oral squamous cell carcinoma
- PC:
-
Pancreatic cancer
- PD-1:
-
Programmed cell death protein 1
- PDK1:
-
Phosphoinositide-dependent kinase-1
- PPI:
-
Protein-protein interactions
- R-2HG:
-
R-2-hydroxyglutarate
- SAM:
-
S-adenosyl-L-methionine
- SKCM:
-
Skin cutaneous melanoma
- STAT3:
-
Signal transducers and activators of transcription 3
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TIME:
-
Tumor immune microenvironment
- TKI:
-
Tyrosine kinase inhibitor
- UGT:
-
UDP-glucuronosyltransferase
- ULK1:
-
Unc-51-like kinase 1
- USP:
-
Ubiquitin-specific protease
- UTR:
-
Untranslated regions
- WTAP:
-
Wilms tumor 1-associated protein
- YAP:
-
Yes-associated protein
- YTH:
-
YT521-B homology
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. https://doi.org/10.3322/caac.21590.
Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer. 2018;124(13):2785–800. https://doi.org/10.1002/cncr.31551.
Maji S, Panda S, Samal SK, Shriwas O, Rath R, Pellecchia M, et al. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv Cancer Res. 2018;137:37–75. https://doi.org/10.1016/bs.acr.2017.11.001.
Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27(Suppl 2):S87–97. https://doi.org/10.3747/co.27.5223.
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. https://doi.org/10.1146/annurev.med.53.082901.103929.
Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med. 2016;22(5):472–8. https://doi.org/10.1038/nm.4091.
Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715. https://doi.org/10.1016/j.drup.2020.100715.
O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67. https://doi.org/10.1038/s41571-018-0142-8.
Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13(10):175. https://doi.org/10.1186/gb-2012-13-10-175.
Adams JM, Cory S. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature. 1975;255(5503):28–33. https://doi.org/10.1038/255028a0.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–5. https://doi.org/10.1073/pnas.71.10.3971.
Beemon K, Keith J. Localization of N6-methyladenosine in the Rous sarcoma virus genome. J Mol Biol. 1977;113(1):165–79. https://doi.org/10.1016/0022-2836(77)90047-x.
Aloni Y, Dhar R, Khoury G. Methylation of nuclear simian virus 40 RNAs. J Virol. 1979;32(1):52–60. https://doi.org/10.1128/JVI.32.1.52-60.1979.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–47.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5. https://doi.org/10.1038/nchembio.1432.
Pendleton KE, Chen B, Liu K, Hunter OV, **e Y, Tu BP, et al. The U6 snRNA m (6) A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169(5):824–35 e14. https://doi.org/10.1016/j.cell.2017.05.003.
** XL, Sun BF, Wang L, **ao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89. https://doi.org/10.1038/cr.2014.3.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–96. https://doi.org/10.1016/j.celrep.2014.05.048.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m (6) A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10. https://doi.org/10.1038/s41421-018-0019-0.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m (6) A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73. https://doi.org/10.1038/nature19342.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 Regulates Nuclear RNA m (6) A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 2018;69(6):1028–38 e6. https://doi.org/10.1016/j.molcel.2018.02.015.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7. https://doi.org/10.1038/nchembio.687.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. https://doi.org/10.1016/j.molcel.2012.10.015.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N (6)-methyladenosine methylated mRNAs. Elife. 2017:6. https://doi.org/10.7554/eLife.31311.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N (6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–27. https://doi.org/10.1038/cr.2017.99.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N (6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161(6):1388–99. https://doi.org/10.1016/j.cell.2015.05.014.
Du H, Zhao Y, He J, Zhang Y, ** H, Liu M, et al. YTHDF2 destabilizes m (6) A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. https://doi.org/10.1038/ncomms12626.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N (6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28. https://doi.org/10.1038/cr.2017.15.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m (6) A-Dependent Nuclear RNA Processing Events. Cell. 2015;162(6):1299–308. https://doi.org/10.1016/j.cell.2015.08.011.
Huang H, Han Y, Zhang C, Wu J, Feng J, Qu L, et al. HNRNPC as a candidate biomarker for chemoresistance in gastric cancer. Tumour Biol. 2016;37(3):3527–34. https://doi.org/10.1007/s13277-015-4144-1.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N (6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95. https://doi.org/10.1038/s41556-018-0045-z.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m (6) A Promotes Cap-Independent Translation. Cell. 2015;163(4):999–1010. https://doi.org/10.1016/j.cell.2015.10.012.
Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74(4):640–50. https://doi.org/10.1016/j.molcel.2019.04.025.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88. https://doi.org/10.1186/s12943-020-01204-7.
Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J, et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer. 2022;21(1):32. https://doi.org/10.1186/s12943-022-01508-w.
Qin S, Mao Y, Wang H, Duan Y, Zhao L. The interplay between m6A modification and non-coding RNA in cancer stemness modulation: mechanisms, signaling pathways, and clinical implications. Int J Biol Sci. 2021;17(11):2718–36. https://doi.org/10.7150/ijbs.60641.
Qu F, Tsegay PS, Liu Y. N (6)-Methyladenosine, DNA Repair, and Genome Stability. Front Mol Biosci. 2021;8:645823. https://doi.org/10.3389/fmolb.2021.645823.
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121. https://doi.org/10.1186/s13045-019-0805-7.
Li X, Ma S, Deng Y, Yi P, Yu J. Targeting the RNA m (6) A modification for cancer immunotherapy. Mol Cancer. 2022;21(1):76. https://doi.org/10.1186/s12943-022-01558-0.
Lan Q, Liu PY, Bell JL, Wang JY, Huttelmaier S, Zhang XD, et al. The Emerging Roles of RNA m (6) A Methylation and Demethylation as Critical Regulators of Tumorigenesis, Drug Sensitivity, and Resistance. Cancer Res. 2021;81(13):3431–40. https://doi.org/10.1158/0008-5472.CAN-20-4107.
Nussinov R, Tsai CJ, Jang H. Anticancer drug resistance: An update and perspective. Drug Resist Updat. 2021;59:100796. https://doi.org/10.1016/j.drup.2021.100796.
Dallavalle S, Dobricic V, Lazzarato L, Gazzano E, Machuqueiro M, Pajeva I, et al. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist Updat. 2020;50:100682. https://doi.org/10.1016/j.drup.2020.100682.
Kathawala RJ, Gupta P, Ashby CR Jr, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. https://doi.org/10.1016/j.drup.2014.11.002.
Li W, Zhang H, Assaraf YG, Zhao K, Xu X, **e J, et al. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. 2016;27:14–29. https://doi.org/10.1016/j.drup.2016.05.001.
Chen Z, Wu L, Zhou J, Lin X, Peng Y, Ge L, et al. N6-methyladenosine-induced ERRgamma triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics. 2020;10(8):3382–96. https://doi.org/10.7150/thno.40144.
Shi Y, Dou Y, Zhang J, Qi J, **n Z, Zhang M, et al. The RNA N6-Methyladenosine Methyltransferase METTL3 Promotes the Progression of Kidney Cancer via N6-Methyladenosine-Dependent Translational Enhancement of ABCD1. Front Cell Dev Biol. 2021;9:737498. https://doi.org/10.3389/fcell.2021.737498.
**ao P, Liu YK, Han W, Hu Y, Zhang BY, Liu WL. Exosomal Delivery of FTO Confers Gefitinib Resistance to Recipient Cells through ABCC10 Regulation in an m6A-dependent Manner. Mol Cancer Res. 2021;19(4):726–38. https://doi.org/10.1158/1541-7786.MCR-20-0541.
Joyce H, McCann A, Clynes M, Larkin A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin Drug Metab Toxicol. 2015;11(5):795–809. https://doi.org/10.1517/17425255.2015.1028356.
Nakano M, Ondo K, Takemoto S, Fukami T, Nakajima M. Methylation of adenosine at the N (6) position post-transcriptionally regulates hepatic P450s expression. Biochem Pharmacol. 2020;171:113697. https://doi.org/10.1016/j.bcp.2019.113697.
Takemoto S, Nakano M, Fukami T, Nakajima M. m (6) A modification impacts hepatic drug and lipid metabolism properties by regulating carboxylesterase 2. Biochem Pharmacol. 2021;193:114766. https://doi.org/10.1016/j.bcp.2021.114766.
Ondo K, Isono M, Nakano M, Hashiba S, Fukami T, Nakajima M. The N (6)-methyladenosine modification posttranscriptionally regulates hepatic UGT2B7 expression. Biochem Pharmacol. 2021;189:114402. https://doi.org/10.1016/j.bcp.2020.114402.
Wang L, Wang H, Song D, Xu M, Liebmen M. New strategies for targeting drug combinations to overcome mutation-driven drug resistance. Semin Cancer Biol. 2017;42:44–51. https://doi.org/10.1016/j.semcancer.2016.11.002.
Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD, et al. An N (6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134–45. https://doi.org/10.1016/j.bcp.2018.12.014.
Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–400. https://doi.org/10.1038/nm.3388.
Bhattarai PY, Kim G, Poudel M, Lim SC, Choi HS. METTL3 induces PLX4032 resistance in melanoma by promoting m (6) A-dependent EGFR translation. Cancer Lett. 2021;522:44–56. https://doi.org/10.1016/j.canlet.2021.09.015.
Huang X, Zhu L, Wang L, Huang W, Tan L, Liu H, et al. YTHDF1 promotes intrahepatic cholangiocarcinoma progression via regulating EGFR mRNA translation. J Gastroenterol Hepatol. 2022. https://doi.org/10.1111/jgh.15816.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61. https://doi.org/10.1016/j.canlet.2018.11.006.
Brinkman JA, Liu Y, Kron SJ. Small-molecule drug repurposing to target DNA damage repair and response pathways. Semin Cancer Biol. 2021;68:230–41. https://doi.org/10.1016/j.semcancer.2020.02.013.
Li H, Wang C, Lan L, Yan L, Li W, Evans I, et al. METTL3 promotes oxaliplatin resistance of gastric cancer CD133+ stem cells by promoting PARP1 mRNA stability. Cell Mol Life Sci. 2022;79(3):135. https://doi.org/10.1007/s00018-022-04129-0.
Narayanan S, Cai CY, Assaraf YG, Guo HQ, Cui Q, Wei L, et al. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist Updat. 2020;48:100663. https://doi.org/10.1016/j.drup.2019.100663.
Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L, et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene. 2017;36(48):6680–90. https://doi.org/10.1038/onc.2017.279.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9. https://doi.org/10.3892/ijo.2017.4219.
Sun Y, Dong D, **a Y, Hao L, Wang W, Zhao C. YTHDF1 promotes breast cancer cell growth, DNA damage repair and chemoresistance. Cell Death Dis. 2022;13(3):230. https://doi.org/10.1038/s41419-022-04672-5.
Wu Y, Wang Z, Han L, Guo Z, Yan B, Guo L, et al. PRMT5 regulates RNA m6A demethylation for doxorubicin sensitivity in breast cancer. Mol Ther. 2022. https://doi.org/10.1016/j.ymthe.2022.03.003.
Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20–33. https://doi.org/10.1038/nrc.2015.2.
Sabnis AJ, Bivona TG. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol Med. 2019;25(3):185–97. https://doi.org/10.1016/j.molmed.2018.12.009.
Allen JE, Prabhu VV, Talekar M, van den Heuvel AP, Lim B, Dicker DT, et al. Genetic and Pharmacological Screens Converge in Identifying FLIP, BCL2, and IAP Proteins as Key Regulators of Sensitivity to the TRAIL-Inducing Anticancer Agent ONC201/TIC10. Cancer Res. 2015;75(8):1668–74. https://doi.org/10.1158/0008-5472.CAN-14-2356.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. https://doi.org/10.1038/s41571-020-0341-y.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N (6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28(11):1062–76. https://doi.org/10.1038/s41422-018-0097-4.
Zhu H, Gan X, Jiang X, Diao S, Wu H, Hu J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J Exp Clin Cancer Res. 2019;38(1):163. https://doi.org/10.1186/s13046-019-1159-2.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076. https://doi.org/10.1016/j.gene.2019.144076.
Wei W, Huo B, Shi X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag Res. 2019;11:1177–87. https://doi.org/10.2147/CMAR.S181058.
Hou H, Zhao H, Yu X, Cong P, Zhou Y, Jiang Y, et al. METTL3 promotes the proliferation and invasion of esophageal cancer cells partly through AKT signaling pathway. Pathol Res Pract. 2020;216(9):153087. https://doi.org/10.1016/j.prp.2020.153087.
Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 Promotes the Proliferation and Mobility of Gastric Cancer Cells. Open Med (Wars). 2019;14:25–31. https://doi.org/10.1515/med-2019-0005.
Adar Y, Stark M, Bram EE, Nowak-Sliwinska P, van den Bergh H, Szewczyk G, et al. Imidazoacridinone-dependent lysosomal photodestruction: a pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012;3:e293. https://doi.org/10.1038/cddis.2012.30.
Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17(23):7337–46. https://doi.org/10.1158/1078-0432.CCR-11-1667.
Piya S, Andreeff M, Borthakur G. Targeting autophagy to overcome chemoresistance in acute myleogenous leukemia. Autophagy. 2017;13(1):214–5. https://doi.org/10.1080/15548627.2016.1245263.
Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33. https://doi.org/10.1016/j.drup.2015.11.004.
Hancock MK, Hermanson SB, Dolman NJ. A quantitative TR-FRET plate reader immunoassay for measuring autophagy. Autophagy. 2012;8(8):1227–44. https://doi.org/10.4161/auto.20441.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m (6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39(12):e103181. https://doi.org/10.15252/embj.2019103181.
** S, Zhang X, Miao Y, Liang P, Zhu K, She Y, et al. m (6) A RNA modification controls autophagy through upregulating ULK1 protein abundance. Cell Res. 2018;28(9):955–7. https://doi.org/10.1038/s41422-018-0069-8.
Wang X, Wu R, Liu Y, Zhao Y, Bi Z, Yao Y, et al. m (6) A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy. 2020;16(7):1221–35. https://doi.org/10.1080/15548627.2019.1659617.
Hao W, Dian M, Zhou Y, Zhong Q, Pang W, Li Z, et al. Autophagy induction promoted by m (6) A reader YTHDF3 through translation upregulation of FOXO3 mRNA. Nat Commun. 2022;13(1):5845. https://doi.org/10.1038/s41467-022-32963-0.
Assaraf YG, Brozovic A, Goncalves AC, Jurkovicova D, Line A, Machuqueiro M, et al. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat. 2019;46:100645. https://doi.org/10.1016/j.drup.2019.100645.
Leonetti A, Assaraf YG, Veltsista PD, El Hassouni B, Tiseo M, Giovannetti E. MicroRNAs as a drug resistance mechanism to targeted therapies in EGFR-mutated NSCLC: Current implications and future directions. Drug Resist Updat. 2019;42:1–11. https://doi.org/10.1016/j.drup.2018.11.002.
Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16(3):280–96. https://doi.org/10.15252/embr.201439949.
Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y, et al. m (6) A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol Cancer. 2020;19(1):3. https://doi.org/10.1186/s12943-019-1128-6.
Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L, et al. N (6)-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating beta-catenin signaling. Mol Cancer. 2020;19(1):163. https://doi.org/10.1186/s12943-020-01281-8.
He JJ, Li Z, Rong ZX, Gao J, Mu Y, Guan YD, et al. m (6) A Reader YTHDC2 Promotes Radiotherapy Resistance of Nasopharyngeal Carcinoma via Activating IGF1R/AKT/S6 Signaling Axis. Front Oncol. 2020;10:1166. https://doi.org/10.3389/fonc.2020.01166.
Liu Z, Lu J, Fang H, Sheng J, Cui M, Yang Y, et al. m6A Modification-Mediated DUXAP8 Regulation of Malignant Phenotype and Chemotherapy Resistance of Hepatocellular Carcinoma Through miR-584-5p/MAPK1/ERK Pathway Axis. Front Cell Dev Biol. 2021;9:783385. https://doi.org/10.3389/fcell.2021.783385.
Ding N, You A, Tian W, Gu L, Deng D. Chidamide increases the sensitivity of Non-small Cell Lung Cancer to Crizotinib by decreasing c-MET mRNA methylation. Int J Biol Sci. 2020;16(14):2595–611. https://doi.org/10.7150/ijbs.45886.
Sun S, Gao T, Pang B, Su X, Guo C, Zhang R, et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m (6) A-dependent manner. Cell Death Dis. 2022;13(1):73. https://doi.org/10.1038/s41419-022-04524-2.
Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25(1):20. https://doi.org/10.1186/s12929-018-0426-4.
Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–67. https://doi.org/10.1016/j.semcancer.2018.11.006.
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, et al. m (6) A modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18(1):185. https://doi.org/10.1186/s12943-019-1116-x.
Liu Z, Wu K, Gu S, Wang W, **e S, Lu T, et al. A methyltransferase-like 14/miR-99a-5p/tribble 2 positive feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2-mediated epigenetic modulation in esophageal squamous cell carcinoma. Clin Transl Med. 2021;11(9):e545. https://doi.org/10.1002/ctm2.545.
Rong D, Wu F, Lu C, Sun G, Shi X, Chen X, et al. m6A modification of circHPS5 and hepatocellular carcinoma progression through HMGA2 expression. Mol Ther Nucleic Acids. 2021;26:637–48. https://doi.org/10.1016/j.omtn.2021.09.001.
Petri BJ, Piell KM, South Whitt GC, Wilt AE, Poulton CC, Lehman NL, et al. HNRNPA2B1 regulates tamoxifen- and fulvestrant-sensitivity and hallmarks of endocrine resistance in breast cancer cells. Cancer Lett. 2021;518:152–68. https://doi.org/10.1016/j.canlet.2021.07.015.
Gu Y, Wu X, Zhang J, Fang Y, Pan Y, Shu Y, et al. The evolving landscape of N (6)-methyladenosine modification in the tumor microenvironment. Mol Ther. 2021;29(5):1703–15. https://doi.org/10.1016/j.ymthe.2021.04.009.
Luo Y, Sun Y, Li L, Mao Y. METTL3 May Regulate Testicular Germ Cell Tumors Through EMT and Immune Pathways. Cell Transplant. 2020;29:963689720946653. https://doi.org/10.1177/0963689720946653.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m (6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39(20):e104514. https://doi.org/10.15252/embj.2020104514.
Li H, Su Q, Li B, Lan L, Wang C, Li W, et al. High expression of WTAP leads to poor prognosis of gastric cancer by influencing tumour-associated T lymphocyte infiltration. J Cell Mol Med. 2020;24(8):4452–65. https://doi.org/10.1111/jcmm.15104.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell. 2020;38(1):79–96 e11. https://doi.org/10.1016/j.ccell.2020.04.017.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m (6) A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10(1):2782. https://doi.org/10.1038/s41467-019-10669-0.
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021;33(6):1221–33 e11. https://doi.org/10.1016/j.cmet.2021.04.001.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117(33):20159–70. https://doi.org/10.1073/pnas.1918986117.
Lin X, Wang Z, Yang G, Wen G, Zhang H. YTHDF2 correlates with tumor immune infiltrates in lower-grade glioma. Aging (Albany NY). 2020;12(18):18476–500. https://doi.org/10.18632/aging.103812.
Tsuchiya K, Yoshimura K, Inoue Y, Iwashita Y, Yamada H, Kawase A, et al. YTHDF1 and YTHDF2 are associated with better patient survival and an inflamed tumor-immune microenvironment in non-small-cell lung cancer. Oncoimmunology. 2021;10(1):1962656. https://doi.org/10.1080/2162402X.2021.1962656.
Su G, Liu T, Han X, Sun H, Che W, Hu K, et al. YTHDF2 is a Potential Biomarker and Associated with Immune Infiltration in Kidney Renal Clear Cell Carcinoma. Front Pharmacol. 2021;12:709548. https://doi.org/10.3389/fphar.2021.709548.
Hu Y, Pan Q, Wang M, Ai X, Yan Y, Tian Y, et al. m (6) A RNA Methylation Regulator YTHDF1 Correlated With Immune Microenvironment Predicts Clinical Outcomes and Therapeutic Efficacy in Breast Cancer. Front Med (Lausanne). 2021;8:667543. https://doi.org/10.3389/fmed.2021.667543.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478). https://doi.org/10.1126/science.aau6977.
Sun Z, Shi K, Yang S, Liu J, Zhou Q, Wang G, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. 2018;17(1):147. https://doi.org/10.1186/s12943-018-0897-7.
Liu T, Li P, Li J, Qi Q, Sun Z, Shi S, et al. Exosomal and intracellular miR-320b promotes lymphatic metastasis in esophageal squamous cell carcinoma. Mol Ther Oncolytics. 2021;23:163–80. https://doi.org/10.1016/j.omto.2021.09.003.
Pan S, Deng Y, Fu J, Zhang Y, Zhang Z, Qin X. N6methyladenosine upregulates miR181d5p in exosomes derived from cancerassociated fibroblasts to inhibit 5FU sensitivity by targeting NCALD in colorectal cancer. Int J Oncol. 2022;60(2). https://doi.org/10.3892/ijo.2022.5304.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276:119399. https://doi.org/10.1016/j.lfs.2021.119399.
**e H, Yao J, Wang Y, Ni B. Exosome-transmitted circVMP1 facilitates the progression and cisplatin resistance of non-small cell lung cancer by targeting miR-524-5p-METTL3/SOX2 axis. Drug Deliv. 2022;29(1):1257–71. https://doi.org/10.1080/10717544.2022.2057617.
Wang Z, He J, Bach DH, Huang YH, Li Z, Liu H, et al. Induction of m (6) A methylation in adipocyte exosomal LncRNAs mediates myeloma drug resistance. J Exp Clin Cancer Res. 2022;41(1):4. https://doi.org/10.1186/s13046-021-02209-w.
Song H, Liu D, Wang L, Liu K, Chen C, Wang L, et al. Methyltransferase like 7B is a potential therapeutic target for reversing EGFR-TKIs resistance in lung adenocarcinoma. Mol Cancer. 2022;21(1):43. https://doi.org/10.1186/s12943-022-01519-7.
Wang T, Liu Z, She Y, Deng J, Zhong Y, Zhao M, et al. A novel protein encoded by circASK1 ameliorates gefitinib resistance in lung adenocarcinoma by competitively activating ASK1-dependent apoptosis. Cancer Lett. 2021;520:321–31. https://doi.org/10.1016/j.canlet.2021.08.007.
Wu Q, Zhang H, Yang D, Min Q, Wang Y, Zhang W, et al. The m6A-induced lncRNA CASC8 promotes proliferation and chemoresistance via upregulation of hnRNPL in esophageal squamous cell carcinoma. Int J Biol Sci. 2022;18(13):4824–36. https://doi.org/10.7150/ijbs.71234.
Huang CS, Zhu YQ, Xu QC, Chen S, Huang Y, Zhao G, et al. YTHDF2 promotes intrahepatic cholangiocarcinoma progression and desensitises cisplatin treatment by increasing CDKN1B mRNA degradation. Clin Transl Med. 2022;12(6):e848. https://doi.org/10.1002/ctm2.848.
Cucciniello L, Gerratana L, Del Mastro L, Puglisi F. Tailoring adjuvant endocrine therapy in early breast cancer: When, how, and how long? Cancer Treat Rev. 2022;110:102445. https://doi.org/10.1016/j.ctrv.2022.102445.
Liu X, Gonzalez G, Dai X, Miao W, Yuan J, Huang M, et al. Adenylate Kinase 4 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an m (6) A-Based Epitranscriptomic Mechanism. Mol Ther. 2020;28(12):2593–604. https://doi.org/10.1016/j.ymthe.2020.09.007.
Li F, Chen S, Yu J, Gao Z, Sun Z, Yi Y, et al. Interplay of m (6) A and histone modifications contributes to temozolomide resistance in glioblastoma. Clin Transl Med. 2021;11(9):e553. https://doi.org/10.1002/ctm2.553.
Li W, Ye K, Li X, Liu X, Peng M, Chen F, et al. YTHDC1 is downregulated by the YY1/HDAC2 complex and controls the sensitivity of ccRCC to sunitinib by targeting the ANXA1-MAPK pathway. J Exp Clin Cancer Res. 2022;41(1):250. https://doi.org/10.1186/s13046-022-02460-9.
Huang H, Weng H, Chen J. m (6) A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37(3):270–88. https://doi.org/10.1016/j.ccell.2020.02.004.
Li S, Jiang F, Chen F, Deng Y, Pan X. Effect of m6A methyltransferase METTL3 -mediated MALAT1/E2F1/AGR2 axis on adriamycin resistance in breast cancer. J Biochem Mol Toxicol. 2022;36(1):e22922. https://doi.org/10.1002/jbt.22922.
** D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. m (6) A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12(1):135. https://doi.org/10.1186/s13045-019-0830-6.
Wei W, Sun J, Zhang H, **ao X, Huang C, Wang L, et al. Circ0008399 Interaction with WTAP Promotes Assembly and Activity of the m (6) A Methyltransferase Complex and Promotes Cisplatin Resistance in Bladder Cancer. Cancer Res. 2021;81(24):6142–56. https://doi.org/10.1158/0008-5472.CAN-21-1518.
Ma H, Shen L, Yang H, Gong H, Du X, Li J. m6A methyltransferase Wilms' tumor 1-associated protein facilitates cell proliferation and cisplatin resistance in NK/T cell lymphoma by regulating dual-specificity phosphatases 6 expression via m6A RNA methylation. IUBMB Life. 2021;73(1):108–17. https://doi.org/10.1002/iub.2410.
Huang T, Cao L, Feng N, Xu B, Dong Y, Wang M. N (6)-methyladenosine (m (6) A)-mediated lncRNA DLGAP1-AS1enhances breast canceradriamycin resistance through miR-299-3p/WTAP feedback loop. Bioengineered. 2021;12(2):10935–44. https://doi.org/10.1080/21655979.2021.2000198.
Li XD, Wang MJ, Zheng JL, Wu YH, Wang X, Jiang XB. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021;112(11):4543–52. https://doi.org/10.1111/cas.15072.
Wang C, Li L, Li M, Wang W, Jiang Z. FTO promotes Bortezomib resistance via m6A-dependent destabilization of SOD2 expression in multiple myeloma. Cancer Gene Ther. 2022. https://doi.org/10.1038/s41417-022-00429-6.
Wang Y, Cheng Z, Xu J, Lai M, Liu L, Zuo M, et al. Fat mass and obesity-associated protein (FTO) mediates signal transducer and activator of transcription 3 (STAT3)-drived resistance of breast cancer to doxorubicin. Bioengineered. 2021;12(1):1874–89. https://doi.org/10.1080/21655979.2021.1924544.
Zhou S, Bai ZL, **a D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57(5):590–7. https://doi.org/10.1002/mc.22782.
Nie S, Zhang L, Liu J, Wan Y, Jiang Y, Yang J, et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J Exp Clin Cancer Res. 2021;40(1):284. https://doi.org/10.1186/s13046-021-02088-1.
Gong H, Liu L, Cui L, Ma H, Shen L. ALKBH5-mediated m6A-demethylation of USP1 regulated T-cell acute lymphoblastic leukemia cell glucocorticoid resistance by Aurora B. Mol Carcinog. 2021;60(9):644–57. https://doi.org/10.1002/mc.23330.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N (6)-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res. 2019;79(11):2812–20. https://doi.org/10.1158/0008-5472.CAN-18-3592.
Shriwas O, Priyadarshini M, Samal SK, Rath R, Panda S, Das Majumdar SK, et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m (6) A-demethylation of FOXM1 and NANOG. Apoptosis. 2020;25(3–4):233–46. https://doi.org/10.1007/s10495-020-01591-8.
Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10(1):4892. https://doi.org/10.1038/s41467-019-12801-6.
Yang Z, Zhao F, Gu X, Feng L, Xu M, Li T, et al. Binding of RNA m6A by IGF2BP3 triggers chemoresistance of HCT8 cells via upregulation of ABCB1. Am J Cancer Res. 2021;11(4):1428–45.
Dong Z, Cui H. The Emerging Roles of RNA Modifications in Glioblastoma. Cancers (Basel). 2020;12(3). https://doi.org/10.3390/cancers12030736.
Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of METTL3 as an Adverse Prognostic Biomarker in Hepatocellular Carcinoma. Dig Dis Sci. 2021;66(4):1110–26. https://doi.org/10.1007/s10620-020-06260-z.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N (6)-methyladenosine (m (6) A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76. https://doi.org/10.1038/nm.4416.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m (6) A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–205. https://doi.org/10.1136/gutjnl-2019-319639.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m (6) A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33. https://doi.org/10.1038/onc.2017.351.
Wang Q, Chen C, Xu X, Shu C, Cao C, Wang Z, et al. APAF1-Binding Long Noncoding RNA Promotes Tumor Growth and Multidrug Resistance in Gastric Cancer by Blocking Apoptosome Assembly. Adv Sci (Weinh). 2022;9(28):e2201889. https://doi.org/10.1002/advs.202201889.
Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28(5):1171–4. https://doi.org/10.1038/leu.2014.16.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19(1):326. https://doi.org/10.1186/s12885-019-5538-z.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N (6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31(1):127–41. https://doi.org/10.1016/j.ccell.2016.11.017.
Zheng QK, Ma C, Ullah I, Hu K, Ma RJ, Zhang N, et al. Roles of N6-Methyladenosine Demethylase FTO in Malignant Tumors Progression. Onco Targets Ther. 2021;14:4837–46. https://doi.org/10.2147/OTT.S329232.
Sun CY, Nie J, Huang JP, Zheng GJ, Feng B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed Pharmacother. 2019;117:109135. https://doi.org/10.1016/j.biopha.2019.109135.
Zhang J, Guo S, Piao HY, Wang Y, Wu Y, Meng XY, et al. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J Physiol Biochem. 2019;75(3):379–89. https://doi.org/10.1007/s13105-019-00690-8.
Yang P, Wang Q, Liu A, Zhu J, Feng J. ALKBH5 Holds Prognostic Values and Inhibits the Metastasis of Colon Cancer. Pathol Oncol Res. 2020;26(3):1615–23. https://doi.org/10.1007/s12253-019-00737-7.
** H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N (6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207. https://doi.org/10.1016/j.ebiom.2019.07.068.
Zhang Y, Liu X, Wang Y, Lai S, Wang Z, Yang Y, et al. The m (6) A demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Mol Cancer. 2022;21(1):174. https://doi.org/10.1186/s12943-022-01647-0.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, et al. MicroRNA-145 Modulates N (6)-Methyladenosine Levels by Targeting the 3′-Untranslated mRNA Region of the N (6)-Methyladenosine Binding YTH Domain Family 2 Protein. J Biol Chem. 2017;292(9):3614–23. https://doi.org/10.1074/jbc.M116.749689.
Zhou L, Jiang J, Huang Z, ** P, Peng L, Luo M, et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/beta-catenin signaling to promote colorectal cancer progression by preventing m (6) A-mediated degradation of STEAP3 mRNA. Mol Cancer. 2022;21(1):168. https://doi.org/10.1186/s12943-022-01638-1.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134(43):17963–71. https://doi.org/10.1021/ja3064149.
Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5(8):658–65. https://doi.org/10.1021/cn500042t.
Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS One. 2016;11(7):e0159072. https://doi.org/10.1371/journal.pone.0159072.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84. https://doi.org/10.1093/nar/gku1276.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m (6) A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18(11):2622–34. https://doi.org/10.1016/j.celrep.2017.02.059.
**ao L, Li X, Mu Z, Zhou J, Zhou P, **e C, et al. FTO Inhibition Enhances the Antitumor Effect of Temozolomide by Targeting MYC-miR-155/23a Cluster-MXI1 Feedback Circuit in Glioma. Cancer Res. 2020;80(18):3945–58. https://doi.org/10.1158/0008-5472.CAN-20-0132.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35(4):677–91 e10. https://doi.org/10.1016/j.ccell.2019.03.006.
Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM. m (6) A-RNA Demethylase FTO Inhibitors Impair Self-Renewal in Glioblastoma Stem Cells. ACS Chem Biol. 2021;16(2):324–33. https://doi.org/10.1021/acschembio.0c00841.
Han X, Wang N, Li J, Wang Y, Wang R, Chang J. Identification of nafamostat mesilate as an inhibitor of the fat mass and obesity-associated protein (FTO) demethylase activity. Chem Biol Interact. 2019;297:80–4. https://doi.org/10.1016/j.cbi.2018.10.023.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m (6) A/MYC/CEBPA Signaling. Cell. 2018;172(1–2):90–105 e23. https://doi.org/10.1016/j.cell.2017.11.031.
Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m (6) A/PFKP/LDHB axis. Mol Cell. 2021;81(5):922–39 e9. https://doi.org/10.1016/j.molcel.2020.12.026.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, Caflisch A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem. 2020;15(9):744–8. https://doi.org/10.1002/cmdc.202000011.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597–601. https://doi.org/10.1038/s41586-021-03536-w.
Moroz-Omori EV, Huang D, Kumar Bedi R, Cheriyamkunnel SJ, Bochenkova E, Dolbois A, et al. METTL3 Inhibitors for Epitranscriptomic Modulation of Cellular Processes. ChemMedChem. 2021;16(19):3035–43. https://doi.org/10.1002/cmdc.202100291.
Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, et al. Discovery of Small Molecules that Activate RNA Methylation through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep. 2019;26(13):3762–71 e5. https://doi.org/10.1016/j.celrep.2019.02.100.
Muller S, Bley N, Busch B, Glass M, Lederer M, Misiak C, et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020;48(15):8576–90. https://doi.org/10.1093/nar/gkaa653.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl Oncol. 2017;10(5):818–27. https://doi.org/10.1016/j.tranon.2017.07.008.
Lan L, Sun YJ, ** XY, **e LJ, Liu L, Cheng L. A Light-Controllable Chemical Modulation of m (6) A RNA Methylation. Angew Chem Int Ed Engl. 2021;60(33):18116–21. https://doi.org/10.1002/anie.202103854.
Song S, Fan G, Li Q, Su Q, Zhang X, Xue X, et al. IDH2 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in multiple myeloma. Oncogene. 2021;40(35):5393–402. https://doi.org/10.1038/s41388-021-01939-7.
Acknowledgments
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
ZQL, XWH, and QD provided direction and guidance throughout the preparation of this manuscript. HJZ, ZQL, and QD wrote and edited the manuscript. QD reviewed and made significant revisions to the manuscript. ZKZ, JXL, HYL, HX, LL, YYZ, QD, and ZQL collected and prepared the related papers. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Z., Zou, H., Dang, Q. et al. Biological and pharmacological roles of m6A modifications in cancer drug resistance. Mol Cancer 21, 220 (2022). https://doi.org/10.1186/s12943-022-01680-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01680-z