
Abstract
Gastric cancer remains a leading cause of cancer-related death worldwide, largely due to inadequate screening methods, late diagnosis, and limited treatment options. Liquid biopsy has emerged as a promising non-invasive approach for cancer screening and prognosis by detecting circulating tumor components like circulating tumor DNA (ctDNA) in the blood. Numerous gastric cancer-specific ctDNA biomarkers have now been identified. CtDNA analysis provides insight into genetic and epigenetic alterations in tumors, holding promise for predicting treatment response and prognosis in gastric cancer patients. This review summarizes current research on ctDNA biology and detection technologies, while highlighting clinical applications of ctDNA for gastric cancer diagnosis, prognosis, and guiding treatment decisions. Current challenges and future perspectives for ctDNA analysis are also discussed.
Similar content being viewed by others


Introduction
Gastric cancer (GC) represents the fifth most common tumor and the fourth leading cause of cancer-related deaths worldwide [1]. According to World Health Organization statistics, the global incidence of GC is increasing continuously, from 1.09 million in 2020 to 1.77 million by 2040 [2]. As early GC is restricted to the mucosa and submucosa, the 5-year survival rate is over 90%. However, the prognosis is poor for advanced GC, with an average survival of only 12 months [3].
The diagnosis of GC is often made at an advanced stage due to the absence of early distinguishable symptoms and the need for a practical mass screening approach for the general population. Although serological tests, including pepsinogen I, pepsinogen II, pepsinogen ratio, gastrin-17, helicobacter pylori antibody, and carbohydrate antigen72-4 (CA72-4) [4], are less invasive, their sensitivity and specificity are limited. The Japanese GC Association has concluded that serum biomarkers are not helpful for early GC diagnosis but can be used to detect recurrence and distant metastases and to predict patient survival and postoperative recurrence [5]. Currently, the mainstay to confirm GC is endoscopy and tissue biopsy, both of which are invasive operations and dependent on the operator's skill. Thus, they are impractical for a mass screening program [6, 7]. Therefore, there is an urgent need for a less invasive, more sensitive, specific, and highly cost-effective test to improve the clinical utility for diagnosis, prognostic assessment, monitoring changes, and guiding treatment options.
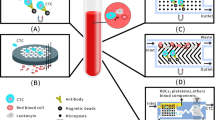
During the past decade, liquid biopsy has become a valuable tool in cancer detection by analyzing tumor-derived entities circulating in body fluids, determining the tissue of origin, monitoring prognosis, and assessing response and resistance to the treatment [8, 9]. These biomarkers include cell-free DNA (cfDNA), cell-free RNA, proteins, autoantibodies, circulating tumor cells, circulating tumor DNA (ctDNA), and cancer-derived extracellular vesicles [10]. Among them, ctDNA is the cornerstone of liquid biopsy in cancer applications due to its intimate relationship with tumors and has become a popular research topic in recent years [11, 12]. In this review, following a brief overview of the biology and detection technologies, we summarized the clinical applications of ctDNA, focusing on its potential in the diagnosis, prognosis, and therapy of GC (Fig. 1).

Clinical applications of liquid biopsy in gastric cancer. Liquid biopsy, including circulating tumor DNA (ctDNA), circulating tumor RNA (ctRNA), extracellular vesicle, and circulating tumor cell (CTC), has gained popularity as a valuable tool in clinical applications of gastric cancer
Circulating tumor DNA
The biological basis of ctDNA
cfDNA, identified by Mandel and Métais in 1948 [13], refers to extracellular DNA found in blood or body fluids, which can be either single-stranded or double-stranded [14]. In healthy individuals, cfDNA is primarily derived from apoptotic or necrotic cells or secreted from lymphocytes and other nucleated cells, which form small homogeneous DNA fragments less than 180 bp in length and 3.6–5.0 ng/mL in concentration. cfDNA has an estimated half-life between 16 min and 2.5 h, depending on factors such as the type and stage of the tumor [15].
In 1977, Leon et al. reported increased cfDNA derived from tumors [16]. After that, Stroun et al. demonstrated that cfDNA contained tumor-related mutations [17]. Therefore, cfDNA derived from tumors is described as ctDNA produced by lysed tumor cells or micrometastatic sites [18]. As a matter of principle, ctDNA contains the same genetic features as the tumor cells, such as single nucleotide mutations and methylation changes [19]. This distinguishes ctDNA from cfDNA and guides the development of cancer detection technologies. Since then, many studies have investigated the potential clinical utility of ctDNA analysis for various cancers [11, 12, 20, 21]. Researchers have gradually realized that the development of ctDNA research holds promise for advances in oncology diagnosis and prognosis prediction.
Advantages and disadvantages of ctDNA testing
Tissue biopsy is currently considered the gold standard for diagnosing and treating cancers. It enables tumor classification, aggressiveness and progression assessment, and genetic composition and mutational phenotype analysis, thereby facilitating personalized treatment strategies [22]. However, ctDNA detection has several advantages over tissue biopsy. Firstly, tissue biopsy is invasive, expensive, and risks complications such as bleeding, local infection, and damage to adjacent tissues [23]. Sometimes, tissue biopsy is not feasible due to anatomic location or underlying coagulation dysfunction. There may also be an increased chance of false negative results due to the limited retrieval of the tumor tissue [24]. In contrast, ctDNA testing requires only a minimum of invasiveness to acquire cancer-related information, regardless of the location of the tumor. Secondly, tissue biopsy only provides information at a specific site and time point. At the same time, blood can be conveniently drawn for ctDNA testing at any time throughout the disease, thus allowing for real-time monitoring of tumor changes without the need for multiple invasive tissue biopsies or imaging surveillance. The short half-life of ctDNA makes it convincing for dynamic monitoring of disease progression [25]. Finally, analysis of ctDNA provides a comprehensive molecular profile of a patient's malignancy, thereby overcoming the challenges posed by intra-tumor heterogeneity and providing additional supportive information in the diagnosis and treatment selection [26].
Despite its potential, ctDNA has several drawbacks that impede its use. Firstly, ctDNA is generally present in low abundance in early-stage cancer and represents only a tiny fraction of total cfDNA (ranging from less than 0.1% to more than 10%), which is further diluted by DNA from non-tumor sources. Currently, detecting tumor-specific mutations on cfDNA is the only way of identifying the ctDNA [19]. Secondly, the proportion of ctDNA in total cfDNA depends on tumor load, cancer stage, cell renewal, and therapy response. It is estimated that patients with a tumor load of 100 g (about 3 × 1010 tumor cells) release 3.3% of their tumor DNA into circulation each day [27]. Hence, ctDNA is frequently undetectable in patients with a low tumor burden or at early stages. Thirdly, ctDNA fragments have a half-life of less than 2 h, requiring rapid processing and stringent pre-analytical procedures such as blood collection, transport, processing, and storage temperatures [28]. Fourthly, there is no consensus on standard experimental procedures for ctDNA assays, including sampling, storage conditions, cfDNA isolation and concentration, data analysis, and interpretation [29], leading to a lack of comparability between studies [30]. Finally, most current clinical studies are retrospective and small in sample size, highlighting an urgent need for multicenter, long-term prospective clinical trials to validate the feasibility of ctDNA in cancer detection, monitoring, and treatment [31].
ctDNA detection methods
Changes in ctDNA in plasma can be detected by quantitative and qualitative (Fig. 2). The former refers to total ctDNA concentration, while the latter refers to DNA aberrations such as single nucleotide mutations and methylation changes [32].

Detection methods of ctDNA in gastric cancer. Quantitative and qualitative changes of ctDNA in plasma provide valuable information for cancer. Quantitative change refers to the total ctDNA level, while qualitative changes include ctDNA mutations and methylation changes
The qualitative analysis of ctDNA can be categorized into two types: targeted and non-targeted [33]. The former is restricted to the detection of single or several biomarkers, focusing on known genetic alterations in primary tumors, such as KRAS (Kirsten rat sarcoma viral oncogene), BRAF (v-Raf murine sarcoma viral oncogene homolog B1), and EGFR (epidermal growth factor receptor) [26]. On the other hand, the non-targeted analysis aims to screen the genome and identifies novel genomic abnormalities, usually through whole genome sequencing (WGS) testing. However, sensitive testing of large target regions is costly, so achieving an appropriate balance between target region size and test sensitivity is essential.
Initially, detecting specific mutations in ctDNA relied on standard quantitative reverse transcription polymerase chain reaction (qPCR). However, due to its limited sensitivity, qPCR was performed mainly in advanced patients with high ctDNA levels [34]. In cases of lower tumor load, where the percentage of ctDNA is significantly lower than 0.1%, digital PCR (dPCR) and droplet-based digital PCR (ddPCR) methods overcome these limitations. For example, Pearson et al. developed a screening tool based on recombinant fibroblast growth factor receptor 2 (FGFR2) ctDNA using ddPCR [35]. In addition, further high-resolution PCR-based methods that have been successfully applied to ctDNA analysis include the BEAMing (beads, emulsion, amplification, and magnetics) [36], ARMS-PCR (Amplification Refractory Mutation System PCR) [37], and COLD-PCR (co-amplification at lower denaturation temperature- PCR) at lower denaturation temperatures [38]. PCR-based technology is faster, less expensive, and highly sensitive, allowing for the detection of tumor-associated mutations at frequencies as low as 0.01% [26]. However, its main drawback is that a single test can detect only one or a few mutations, limiting its ability to study significant numbers and different kinds of genomic alterations [39]. In 2018, Cohen et al. developed a PCR-based test, CancerSEEK, and investigated its utility for the early detection of eight common cancers. The results showed that it could be used to assess cancer-specific characteristics in the early stages (I-III) of more than 82% of cancers [40].
Compared to PCR-based methods, next-generation sequencing (NGS)-based technology is characterized by high throughput, high sensitivity, and extensive coverage. It can identify somatic and germline mutations, copy number alterations, and other chromosomal rearrangements, including translocation, conversion, and inversion. Unlike targeted analysis, NGS does not require prior knowledge of the exact genetic changes in tumors, making it a non-targeted approach. Currently, targeted deep sequencing methods include TAM-Seq (tagged-amplicon deep sequencing) [41], Safe-SeqS (Safe-Sequencing) [42], and CAPP-Seq (Computer Aided Process Planning sequencing) [43]. These technologies allow NGS to provide personalized cancer genetic profiles and facilitate personalized medicine [19]. Based on this, Kato et al. demonstrated the feasibility of NGS for ctDNA evaluation in patients with gastroesophageal adenocarcinoma [44].
Whole exome sequencing (WES) and WGS can detect tumor mutations in all patients, making them ideal for genome-wide copy number analysis and detection of significant structural variants. However, their high cost renders them unsuitable for sensitively detecting single nucleotide variants [26]. Despite lower analytical sensitivity for ctDNA analysis throughout the disease course, WES and WGS can track clonal genomic evolution associated with tumor progression [45]. Li et al. developed fingerprinting profiles based on WES for ctDNA in individual patients. This study demonstrated that ctDNA fingerprinting improves the specificity of several tumor types for monitoring treatment response and sensitivity [46].
Although tumor-associated gene mutations have been the focus of biomarker research for a long time, their wide diversity has always been a challenge for develo** validated biomarkers. To achieve sufficient sensitivity, a significant proportion of genomes must be examined [47]. In contrast, epigenetic alterations appear more stable and homogeneous in cancer, making them a promising alternative for biomarker development [48]. DNA methylation is the most widely studied epigenetic modification [49, 50]. There are two main types of methods for detecting ctDNA methylation, namely bisulfite-based conversion methods and non-bisulfite–based conversion methods. The latter includes restriction enzyme-based methods such as methylation-sensitive restriction enzymes (MSREs) [51], enrichment/immune-precipitation-based techniques such as methylated DNA immunoprecipitation sequencing (MeDIP-seq) [52], and 5-hydroxymethylation profiling. Many methylation detection methods based on bisulfite conversion have been developed, such as whole genome bisulfite sequencing (WGBS), reduced-representation bisulfite sequencing (RRBS), methylated CpG tandems amplification and sequencing (MCTA-seq), and methylation arrays [53].
Clinical applications
Diagnosis and screening
In the last few years, we have witnessed a growing body of clinical evidence supporting the detection of cfDNA for screening and monitoring patients with GC (Table 1). This test would be detected four years earlier than the current “gold standard” [54]. Plasma cfDNA levels in cancer patients, including GC patients, are two to three times higher than in healthy individuals [55]. However, plasma cfDNA levels may also increase in response to infection, inflammation, and other stressful conditions [56]. Therefore, quantifying plasma cfDNA would not be a sufficient biomarker to detect cancer due to its lack of specificity.
Information on tumor-associated genetic variants can be detected in ctDNA, ranging from simple point mutations to complex structural variants and even chromosomal copy number variants [57]. Therefore, detecting tumor-associated mutations in ctDNA can provide more identification of GC and guide its detection. Bettegowda et al. [58] first caught ctDNA containing tumor-specific single nucleotide variants in the plasma of 15 GC patients. Following this, Fang et al. [59] analyzed eight genetic alterations and found that Tumor Protein 53 (TP53), AT-Rich Interaction Domain 1A (ARID1A), and phosphatidylinositol-3-kinase catalytic subunit α (PI3KCA) were the most frequently mutated in ctDNA of patients with advanced GC. The detection rate of ctDNA was found to correlate with the tumor stage. Tumor-specific TP53 mutations were detected in patients with stage III-IV GC but not in patients with stage II GC [60]. It has been demonstrated that the copy number of Human epidermal growth factor receptor-2 (HER2) in the plasma of GC patients is significantly higher than that of healthy controls [61]. However, Kinugasa et al. [62] found low concordance between HER2 levels in tumor tissue and plasma DNA. This discordance may be caused by intra-tumor heterogeneity or sampling error due to low ctDNA levels [26]. To investigate whether ctDNA can cover tumor heterogeneity, Gao et al. [63] performed paired sequencing of tumor tissue biopsies and plasma samples from five patients. The biopsies confirmed the presence of tumor heterogeneity, but ctDNA only partially covered this heterogeneity. These analyses suggest that ctDNA research may be superior to tissue biopsy when examining GC with extensive intra-tumor heterogeneity.
In addition to single nucleotide variants, many studies have evaluated ctDNA methylation as a potential biomarker for cancer detection. It has been suggested that epigenetic alterations often precede somatic mutations and are more common than previously thought [64]. Circulating cfDNA methylation is highly predictive for GC, compared to methylation biomarkers in tissues [65]. Hypermethylation of p16 and E-calmodulin gene promoter regions has been detected in serum DNA samples from GC patients but not in healthy volunteers [66]. However, the reported ratio of ctDNA p16 promoter methylation in GC varies significantly across different studies [67], indicating the need for further validation. Ras association domain family 1, form A (RASSF1A), and protocadherin 10 (PCDH10) are tumor suppressor genes. Hypermethylation of RASS1A and PCDH10 was detectable in plasma samples from GC patients [68]. The study by Bernal et al. [69] confirmed the high frequency of methylation of seven genes in GC plasma, including Adenomatous Polyposis Coli (APC), SH2 domain-containing protein tyrosine phosphatase 1 (SHP1), E-calmodulin, Estrogen receptor (ER), Reprimo, Semaphorin-3B (SEMA3B) and 3-O-sulfotransferase-2 (3OST2). Additionally, methylation of tissue factor pathway inhibitor 2 (TFPI2) [70], XIAP associated factor 1 (XAF1) [71], Reprimo-like (RPRML) [72], multiple tumor suppressor 1 (MTS) and Cadherin 1 (CDH1) promoter region [10] dedicator of cytokinesis 10 (DOCK10), calcineurin binding protein 1 (CABIN1) and KQT-like subfamily, member 5 (KCNQ5) [73] can all be used as potential non-invasive diagnostic indicators in GC. In a meta-analysis of 16 studies, Gao et al. [26]. Postoperative tumor-informed ctDNA detection in EGC is feasible and allows for enhanced patient risk stratification and prognostication during curative-intent therapy [94]. GC patients with high ctDNA mutation abundance exhibited shorter overall survival (OS) than those with low mutation abundance [95]. Reduced ctDNA mutation frequency after treatment was associated with improved PFS and OS [96]. Patients with peritoneal metastases have more ctDNA mutated genes than non-peritoneal metastases. Mutations in cell division cycle 27 (CDC27) are associated with a higher risk of peritoneal metastases and a lower survival rate [97]. Patients with Mesenchymal-epithelial transition (MET) amplification in ctDNA have shorter OS than those without MET amplification, which indicates that ctDNA can predict disease progression in patients with advanced GC [98]. In some patients with Epstein-Barr virus (EBV)-associated GC, circulating EBV DNA is reduced after surgery and increases before clinically detectable recurrence. This could help monitor tumor load in patients with EBV-associated GC and predict recurrence [99]. HER2 alterations in ctDNA were significantly associated with poor OS [44]. Patients who tested positive for HER2 ctDNA before treatment had significantly shorter survival than those with negative. Still, no difference in survival was found when comparing the survival of patients regardless of tissue HER2 status [62]. This may be due to tumor heterogeneity, but ctDNA testing may provide a more accurate assessment. Based on a special NGS panel, the number of ctDNA mutations before the start of first-line chemotherapy has prognostic value. Moreover, residual ctDNA after three cycles of systemic treatment is associated with an inferior survival [100]. Changes in genomic features of ctDNA could be biomarkers for predicting the response of platinum-based first-line chemotherapy in patients with advanced GC [28].
Analysis of ctDNA has been shown to provide information on mutations that are not found in tissue biopsies due to intra-tumor heterogeneity, which can help stratify patients for testing targeted drugs and may also help identify new therapeutic targets. Moreover, most current clinical studies on ctDNA are retrospective, with small sample sizes. All these must be explored in more multicenter and long-term prospective clinical trials [26].
Conclusions
GC remains one of the most common malignancies worldwide with a poor prognosis, primarily due to the lack of population-appropriate screening, early detection methods, and suitable treatment options. The application of ctDNA as a biomarker is an exciting and emerging area for disease screening and monitoring in GC. Moreover, combining ctDNA with other biomarkers is expected to enhance cancer management for GC patients in the near future.
Availability of data and materials
Not applicable.
Abbreviations
- CA:
-
Carbohydrate antigen
- CEA:
-
Carcinoembryonic antigen
- cfDNA:
-
Cell-free DNA
- ctDNA:
-
Circulating tumor DNA
- DFS:
-
Disease-free survival
- EBV:
-
Epstein-Barr virus
- FGFR2:
-
Fibroblast growth factor receptor 2
- GC:
-
Gastric cancer
- HER2:
-
Human epidermal growth factor receptor-2
- MET:
-
Mesenchymal-epithelial transition
- OS:
-
Overall survival
- PCDH10:
-
Protocadherin 10
- PCR:
-
Polymerase chain reaction
- PFS:
-
Progression-free survival
- RASSF1A:
-
RAS association domain family 1, form A
- RUNX3:
-
Runt-related transcription factor 3
- TP53:
-
Tumor Protein 53
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Adeniji N, Dhanasekaran R. Current and emerging tools for hepatocellular carcinoma surveillance. Hepatol Commun. 2021;5(12):1972–86.
Rosati G, Ferrara D, Manzione L. New perspectives in the treatment of advanced or metastatic gastric cancer. World J Gastroenterol. 2009;15(22):2689–92.
Huang YK, Yu JC, Kang WM, Ma ZQ, Ye X, Tian SB, Yan C. Significance of serum pepsinogens as a biomarker for gastric cancer and atrophic gastritis screening: a systematic review and meta-analysis. PLoS ONE. 2015;10(11): e0142080.
Shimada H, Noie T, Ohashi M, Oba K, Takahashi Y. Clinical significance of serum tumor markers for gastric cancer: a systematic review of literature by the Task Force of the Japanese Gastric Cancer Association. Gastric Cancer. 2014;17(1):26–33.
Gupta N, Bansal A, Wani SB, Gaddam S, Rastogi A, Sharma P. Endoscopy for upper GI cancer screening in the general population: a cost-utility analysis. Gastrointest Endosc. 2011;74(3):610-624.e612.
Pasechnikov V, Chukov S, Fedorov E, Kikuste I, Leja M. Gastric cancer: prevention, screening and early diagnosis. World J Gastroenterol. 2014;20(38):13842–62.
Lengyel CG, Hussain S, Trapani D, El Bairi K, Altuna SC, Seeber A, Odhiambo A, Habeeb BS, Seid F. The emerging role of liquid biopsy in gastric cancer. J Clin Med. 2021;10(10):2108.
Cellini F, Morganti AG, Di Matteo FM, Mattiucci GC, Valentini V. Clinical management of gastroesophageal junction tumors: past and recent evidences for the role of radiotherapy in the multidisciplinary approach. Radiat Oncol. 2014;9:45.
Huang ZB, Zhang HT, Yu B, Yu DH. Cell-free DNA as a liquid biopsy for early detection of gastric cancer. Oncol Lett. 2021;21(1):3.
Dasari A, Morris VK, Allegra CJ, Atreya C, Benson AB 3rd, Boland P, Chung K, Copur MS, Corcoran RB, Deming DA, et al. ctDNA applications and integration in colorectal cancer: an NCI Colon and Rectal-Anal Task Forces whitepaper. Nat Rev Clin Oncol. 2020;17(12):757–70.
Pantel K, Alix-Panabières C. Liquid biopsy in 2016: Circulating tumour cells and cell-free DNA in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2017;14(2):73–4.
Mandel P, Metais P. Nuclear acids in human blood plasma. C R Seances Soc Biol Fil. 1948;142(3–4):241–3.
Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
Yao W, Mei C, Nan X, Hui L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: a qualitative study. Gene. 2016;590(1):142–8.
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37(3):646–50.
Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46(5):318–22.
Qi Q, Pan YF, Shen JJ, Gu XQ, Han SW, Liao HH, Jiang YZ, Zhong LP. Circulating DNA for detection of gastric cancer. Eur Rev Med Pharmacol Sci. 2016;20(12):2558–64.
Ye Q, Ling S, Zheng S, Xu X. Liquid biopsy in hepatocellular carcinoma: circulating tumor cells and circulating tumor DNA. Mol Cancer. 2019;18(1):114.
Li W, Liu JB, Hou LK, Yu F, Zhang J, Wu W, Tang XM, Sun F, Lu HM, Deng J, et al. Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring. Mol Cancer. 2022;21(1):25.
Papakonstantinou A, Gonzalez NS, Pimentel I, Suñol A, Zamora E, Ortiz C, Espinosa-Bravo M, Peg V, Vivancos A, Saura C, et al. Prognostic value of ctDNA detection in patients with early breast cancer undergoing neoadjuvant therapy: a systematic review and meta-analysis. Cancer Treat Rev. 2022;104: 102362.
Jung A, Kirchner T. Liquid biopsy in tumor genetic diagnosis. Dtsch Arztebl Int. 2018;115(10):169–74.
Sawada K, Kotani D, Bando H. The clinical landscape of circulating tumor DNA in gastrointestinal malignancies. Front Oncol. 2018;8:263.
Sholl LM, Aisner DL, Allen TC, Beasley MB, Cagle PT, Capelozzi VL, Dacic S, Hariri LP, Kerr KM, Lantuejoul S, et al. Liquid biopsy in lung cancer: a perspective from members of the pulmonary pathology society. Arch Pathol Lab Med. 2016;140(8):825–9.
Hofman P, Heeke S, Alix-Panabières C, Pantel K. Liquid biopsy in the era of immuno-oncology: is it ready for prime-time use for cancer patients? Ann Oncol. 2019;30(9):1448–59.
Koldby KM, Mortensen MB, Detlefsen S, Pfeiffer P, Thomassen M, Kruse TA. Tumor-specific genetic aberrations in cell-free DNA of gastroesophageal cancer patients. J Gastroenterol. 2019;54(2):108–21.
Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA Jr, Goodman SN, David KA, Juhl H, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. 2005;102(45):16368–73.
Pessoa LS, Heringer M, Ferrer VP. ctDNA as a cancer biomarker: a broad overview. Crit Rev Oncol Hematol. 2020;155: 103109.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88.
Wu HC, Yang HI, Wang Q, Chen CJ, Santella RM. Plasma DNA methylation marker and hepatocellular carcinoma risk prediction model for the general population. Carcinogenesis. 2017;38(10):1021–8.
Siravegna G, Mussolin B, Venesio T, Marsoni S, Seoane J, Dive C, Papadopoulos N, Kopetz S, Corcoran RB, Siu LL, Bardelli A. How liquid biopsies can change clinical practice in oncology. Ann Oncol. 2019;30(10):1580–90.
Yin CQ, Yuan CH, Qu Z, Guan Q, Chen H, Wang FB. Liquid biopsy of hepatocellular carcinoma: circulating tumor-derived biomarkers. Dis Markers. 2016;2016:1427849.
Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61(1):112–23.
Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–209.
Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, Kilgour E, Smith NR, Geh C, Rooney C, et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 2016;6(8):838–51.
Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3(7):551–9.
Feng Q, Yang ZY, Zhang JT, Tang JL. Comparison of direct sequencing and amplification refractory mutation system for detecting epidermal growth factor receptor mutation in non-small-cell lung cancer patients: a systematic review and meta-analysis. Oncotarget. 2017;8(35):59552–62.
Mauger F, How-Kit A, Tost J. COLD-PCR technologies in the area of personalized medicine: methodology and applications. Mol Diagn Ther. 2017;21(3):269–83.
Xu S, Lou F, Wu Y, Sun DQ, Zhang JB, Chen W, Ye H, Liu JH, Wei S, Zhao MY, et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett. 2016;370(2):324–31.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–30.
Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136): 136ra168.
Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(23):9530–5.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54.
Kato S, Okamura R, Baumgartner JM, Patel H, Leichman L, Kelly K, Sicklick JK, Fanta PT, Lippman SM, Kurzrock R. Analysis of circulating tumor DNA and clinical correlates in patients with esophageal, gastroesophageal junction, and gastric adenocarcinoma. Clin Cancer Res. 2018;24(24):6248–56.
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12.
Li J, Jiang W, Wei J, Zhang J, Cai L, Luo M, Wang Z, Sun W, Wang S, Wang C, et al. Patient specific circulating tumor DNA fingerprints to monitor treatment response across multiple tumors. J Transl Med. 2020;18(1):293.
Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109–27.
Warton K, Samimi G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front Mol Biosci. 2015;2:13.
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92.
Liu Z, Wang Z, Jia E, Ouyang T, Pan M, Lu J, Ge Q, Bai Y. Analysis of genome-wide in cell free DNA methylation: progress and prospect. Analyst. 2019;144(20):5912–22.
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37(8):853–62.
Werner B, Yuwono NL, Henry C, Gunther K, Rapkins RW, Ford CE, Warton K. Circulating cell-free DNA from plasma undergoes less fragmentation during bisulfite treatment than genomic DNA due to low molecular weight. PLoS ONE. 2019;14(10): e0224338.
Chen X, Gole J, Gore A, He Q, Lu M, Min J, Yuan Z, Yang X, Jiang Y, Zhang T, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun. 2020;11(1):3475.
De Mattos-Arruda L, Olmos D, Tabernero J. Prognostic and predictive roles for circulating biomarkers in gastrointestinal cancer. Future Oncol. 2011;7(12):1385–97.
Lui YY, Chik KW, Chiu RW, Ho CY, Lam CW, Lo YM. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48(3):421–7.
Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12(7): e1006162.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224): 224ra224.
Fang WL, Lan YT, Huang KH, Liu CA, Hung YP, Lin CH, Jhang FY, Chang SC, Chen MH, Chao Y, et al. Clinical significance of circulating plasma DNA in gastric cancer. Int J Cancer. 2016;138(12):2974–83.
Hamakawa T, Kukita Y, Kurokawa Y, Miyazaki Y, Takahashi T, Yamasaki M, Miyata H, Nakajima K, Taniguchi K, Takiguchi S, et al. Monitoring gastric cancer progression with circulating tumour DNA. Br J Cancer. 2015;112(2):352–6.
Park KU, Lee HE, Nam SK, Nam KH, Park DJ, Kim HH, Kim WH, Lee HS. The quantification of HER2 and MYC gene fragments in cell-free plasma as putative biomarkers for gastric cancer diagnosis. Clin Chem Lab Med. 2014;52(7):1033–40.
Kinugasa H, Nouso K, Tanaka T, Miyahara K, Morimoto Y, Dohi C, Matsubara T, Okada H, Yamamoto K. Droplet digital PCR measurement of HER2 in patients with gastric cancer. Br J Cancer. 2015;112(10):1652–5.
Gao J, Wang H, Zang W, Li B, Rao G, Li L, Yu Y, Li Z, Dong B, Lu Z, et al. Circulating tumor DNA functions as an alternative for tissue to overcome tumor heterogeneity in advanced gastric cancer. Cancer Sci. 2017;108(9):1881–7.
Weisenberger DJ. Characterizing DNA methylation alterations from the cancer genome atlas. J Clin Invest. 2014;124(1):17–23.
Li W, Zhang X, Lu X, You L, Song Y, Luo Z, Zhang J, Nie J, Zheng W, Xu D, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27(10):1243–57.
Ichikawa D, Koike H, Ikoma H, Ikoma D, Tani N, Otsuji E, Kitamura K, Yamagishi H. Detection of aberrant methylation as a tumor marker in serum of patients with gastric cancer. Anticancer Res. 2004;24(4):2477–81.
Abbaszadegan MR, Moaven O, Sima HR, Ghafarzadegan K, A’Rabi A, Forghani MN, Raziee HR, Mashhadinejad A, Jafarzadeh M, Esmaili-Shandiz E, Dadkhah E. p16 promoter hypermethylation: a useful serum marker for early detection of gastric cancer. World J Gastroenterol. 2008;14(13):2055–60.
Pimson C, Ekalaksananan T, Pientong C, Promthet S, Putthanachote N, Suwanrungruang K, Wiangnon S. Aberrant methylation of PCDH10 and RASSF1A genes in blood samples for non-invasive diagnosis and prognostic assessment of gastric cancer. PeerJ. 2016;4: e2112.
Bernal C, Aguayo F, Villarroel C, Vargas M, Díaz I, Ossandon FJ, Santibáñez E, Palma M, Aravena E, Barrientos C, Corvalan AH. Reprimo as a potential biomarker for early detection in gastric cancer. Clin Cancer Res. 2008;14(19):6264–9.
Hibi K, Goto T, Shirahata A, Saito M, Kigawa G, Nemoto H, Sanada Y. Detection of TFPI2 methylation in the serum of gastric cancer patients. Anticancer Res. 2011;31(11):3835–8.
Ling ZQ, Lv P, Lu XX, Yu JL, Han J, Ying LS, Zhu X, Zhu WY, Fang XH, Wang S, Wu YC. Circulating methylated XAF1 DNA indicates poor prognosis for gastric cancer. PLoS ONE. 2013;8(6): e67195.
Alarcón MA, Olivares W, Córdova-Delgado M, Muñoz-Medel M, de Mayo T, Carrasco-Aviño G, Wichmann I, Landeros N, Amigo J, Norero E, et al. The reprimo-like gene is an epigenetic-mediated tumor suppressor and a candidate biomarker for the non-invasive detection of gastric cancer. Int J Mol Sci. 2020;21(24):9472.
Ren J, Lu P, Zhou X, Liao Y, Liu X, Li J, Wang W, Wang J, Wen L, Fu W, Tang F. Genome-scale methylation analysis of circulating cell-free DNA in gastric cancer patients. CLIN CHEM. 2021;68(2):354–64.
Gao Y, Zhang K, ** H, Cai A, Wu X, Cui J, Li J, Qiao Z, Wei B, Chen L. Diagnostic and prognostic value of circulating tumor DNA in gastric cancer: a meta-analysis. Oncotarget. 2017;8(4):6330–40.
Lu XX, Yu JL, Ying LS, Han J, Wang S, Yu QM, Wang XB, Fang XH, Ling ZQ. Stepwise cumulation of RUNX3 methylation mediated by Helicobacter pylori infection contributes to gastric carcinoma progression. Cancer. 2012;118(22):5507–17.
Sakakura C, Hamada T, Miyagawa K, Nishio M, Miyashita A, Nagata H, Ida H, Yazumi S, Otsuji E, Chiba T, et al. Quantitative analysis of tumor-derived methylated RUNX3 sequences in the serum of gastric cancer patients. Anticancer Res. 2009;29(7):2619–25.
Yan H, Chen W, Ge K, Mao X, Li X, Liu W, Wu J. Value of plasma methylated SFRP2 in prognosis of gastric cancer. Dig Dis Sci. 2020;66:3854.
Lin Z, Luo M, Chen X, He X, Qian Y, Lai S, Si J, Chen S. Combined detection of plasma ZIC1, HOXD10 and RUNX3 methylation is a promising strategy for early detection of gastric cancer and precancerous lesions. J Cancer. 2017;8(6):1038–44.
Kolesnikova EV, Tamkovich SN, Bryzgunova OE, Shelestyuk PI, Permyakova VI, Vlassov VV, Tuzikov AS, Laktionov PP, Rykova EY. Circulating DNA in the blood of gastric cancer patients. Ann N Y Acad Sci. 2008;1137:226–31.
Saluja H, Karapetis CS, Pedersen SK, Young GP, Symonds EL. The use of circulating tumor DNA for prognosis of gastrointestinal cancers. Front Oncol. 2018;8:275.
Tas F, Faruk Aykan N, Aydiner A, Yasasever V, Topuz E. Measurement of serum CA 19–9 may be more valuable than CEA in prediction of recurrence in patients with gastric cancer. Am J Clin Oncol. 2001;24(2):148–9.
Aloe S, D’Alessandro R, Spila A, Ferroni P, Basili S, Palmirotta R, Carlini M, Graziano F, Mancini R, Mariotti S, et al. Prognostic value of serum and tumor tissue CA 72–4 content in gastric cancer. Int J Biol Markers. 2003;18(1):21–7.
Nakamura Y, Taniguchi H, Ikeda M, Bando H, Kato K, Morizane C, Esaki T, Komatsu Y, Kawamoto Y, Takahashi N, et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med. 2020;26(12):1859–64.
Min L, Chen J, Yu M, Yang K, Liu D. Utilizing circulating tumor DNA as a prognostic predictor of gastric cancer: a meta-analysis. Biomarkers. 2023;28:1–20.
Kim K, Shin DG, Park MK, Baik SH, Kim TH, Kim S, Lee S. Circulating cell-free DNA as a promising biomarker in patients with gastric cancer: diagnostic validity and significant reduction of cfDNA after surgical resection. Ann Surg Treat Res. 2014;86(3):136–42.
Pu WY, Zhang R, **ao L, Wu YY, Gong W, Lv XD, Zhong FY, Zhuang ZX, Bai XM, Li K, **ng CG. Prediction of cancer progression in a group of 73 gastric cancer patients by circulating cell-free DNA. BMC Cancer. 2016;16(1):943.
Lan YT, Chen MH, Fang WL, Hsieh CC, Lin CH, Jhang FY, Yang SH, Lin JK, Chen WS, Jiang JK, et al. Clinical relevance of cell-free DNA in gastrointestinal tract malignancy. Oncotarget. 2017;8(2):3009–17.
Kim YW, Kim YH, Song Y, Kim HS, Sim HW, Poojan S, Eom BW, Kook MC, Joo J, Hong KM. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Exp Mol Med. 2019;51(8):1–10.
Normando SRC, Delgado PO, Rodrigues A, David Filho WJ, Fonseca FLA, Cruz F, Del Giglio A. Circulating free plasma tumor DNA in patients with advanced gastric cancer receiving systemic chemotherapy. BMC Clin Pathol. 2018;18:12.
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, Liu XQ, Sher X, Jung H, Lee M, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449–58.
Suzuki T, Suzuki T, Yoshimura Y, Yahata M, Yew PY, Nakamura T, Nakamura Y, Park JH, Matsuo R. Detection of circulating tumor DNA in patients of operative colorectal and gastric cancers. Oncotarget. 2020;11(34):3198–207.
Openshaw MR, Mohamed AA, Ottolini B, Fernandez-Garcia D, Richards CJ, Page K, Guttery DS, Thomas AL, Shaw JA. Longitudinal monitoring of circulating tumour DNA improves prognostication and relapse detection in gastroesophageal adenocarcinoma. Br J Cancer. 2020;123(8):1271–9.
Cabel L, Decraene C, Bieche I, Pierga JY, Bennamoun M, Fuks D, Ferraz JM, Lefevre M, Baulande S, Bernard V, et al. Limited sensitivity of circulating tumor DNA detection by droplet digital PCR in non-metastatic operable gastric cancer patients. Cancers (Basel). 2019;11(3):396.
Huffman BM, Aushev VN, Budde GL, Chao J, Dayyani F, Hanna D, Botta GP, Catenacci DVT, Maron SB, Krinshpun S, et al. Analysis of circulating tumor DNA to predict risk of recurrence in patients with esophageal and gastric cancers. JCO Precis Oncol. 2022;6: e2200420.
Wu R, Shi C, Chen Q, Wu F, Li Q. Detection of circulating tumor cell DNA for monitoring advanced gastric cancer. Int J Clin Exp Pathol. 2020;13(2):203–11.
Qiao G, Wang X, Zhou L, Zhou X, Song Y, Wang S, Zhao L, Morse MA, Hobeika A, Song J, et al. Autologous dendritic cell-cytokine induced killer cell immunotherapy combined with S-1 plus cisplatin in patients with advanced gastric cancer: a prospective study. Clin Cancer Res. 2019;25(5):1494–504.
Wu R, Li Q, Wu F, Shi C, Chen Q. Comprehensive analysis of CDC27 related to peritoneal metastasis by whole exome sequencing in gastric cancer. Onco Targets Ther. 2020;13:3335–46.
Li J, Li Z, Ding Y, Xu Y, Zhu X, Cao N, Huang C, Qin M, Liu F, Zhao A. TP53 mutation and MET amplification in circulating tumor DNA analysis predict disease progression in patients with advanced gastric cancer. PeerJ. 2021;9: e11146.
Shoda K, Ichikawa D, Fujita Y, Masuda K, Hiramoto H, Hamada J, Arita T, Konishi H, Kosuga T, Komatsu S, et al. Clinical utility of circulating cell-free Epstein-Barr virus DNA in patients with gastric cancer. Oncotarget. 2017;8(17):28796–804.
van Velzen MJM, Creemers A, van den Ende T, Schokker S, Krausz S, Reinten RJ, Dijk F, van Noesel CJM, Halfwerk H, Meijer SL, et al. Circulating tumor DNA predicts outcome in metastatic gastroesophageal cancer. Gastric Cancer. 2022;25(5):906–15.
** W, Zhou C, Xu F, Sun D, Wang S, Chen Y, Ji J, Ma T, Wu J, Shangguan C, et al. Molecular evolutionary process of advanced gastric cancer during sequential chemotherapy detected by circulating tumor DNA. J Transl Med. 2022;20(1):365.
Tani N, Ichikawa D, Ikoma D, Ikoma H, Sai S, Tomita H, Okamoto K, Kikuchi S, Fujiwara H, Ochiai T, et al. An early detection of recurrence using reverse transcriptase-polymerase chain reaction (RT-PCR) and methylation-specific polymerase chain reaction (MSP) from peripheral blood in patients after gastrectomy. Gan To Kagaku Ryoho. 2006;33(12):1720–2.
Han J, Lv P, Yu JL, Wu YC, Zhu X, Hong LL, Zhu WY, Yu QM, Wang XB, Li P, Ling ZQ. Circulating methylated MINT2 promoter DNA is a potential poor prognostic factor in gastric cancer. Dig Dis Sci. 2014;59(6):1160–8.
Hu XY, Ling ZN, Hong LL, Yu QM, Li P, Ling ZQ. Circulating methylated THBS1 DNAs as a novel marker for predicting peritoneal dissemination in gastric cancer. J Clin Lab Anal. 2021;35(9): e23936.
Balgkouranidou I, Matthaios D, Karayiannakis A, Bolanaki H, Michailidis P, Xenidis N, Amarantidis K, Chelis L, Trypsianis G, Chatzaki E, et al. Prognostic role of APC and RASSF1A promoter methylation status in cell free circulating DNA of operable gastric cancer patients. Mutat Res. 2015;778:46–51.
Karamitrousis EI, Balgkouranidou I, Xenidis N, Amarantidis K, Biziota E, Koukaki T, Trypsianis G, Karayiannakis A, Bolanaki H, Kolios G, et al. Prognostic role of RASSF1A, SOX17 and Wif-1 promoter methylation status in cell-free DNA of advanced gastric cancer patients. Technol Cancer Res Treat. 2021;20:1533033820973279.
Yu JL, Lv P, Han J, Zhu X, Hong LL, Zhu WY, Wang XB, Wu YC, Li P, Ling ZQ. Methylated TIMP-3 DNA in body fluids is an independent prognostic factor for gastric cancer. Arch Pathol Lab Med. 2014;138(11):1466–73.
Takahashi T, Saikawa Y, Kitagawa Y. Gastric cancer: current status of diagnosis and treatment. Cancers (Basel). 2013;5(1):48–63.
Nakamura Y, Kawazoe A, Lordick F, Janjigian YY, Shitara K. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: an emerging paradigm. Nat Rev Clin Oncol. 2021;18:473.
Shoda K, Ichikawa D, Fujita Y, Masuda K, Hiramoto H, Hamada J, Arita T, Konishi H, Komatsu S, Shiozaki A, et al. Monitoring the HER2 copy number status in circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer. 2017;20(1):126–35.
Wang H, Li B, Liu Z, Gong J, Shao L, Ren J, Niu Y, Bo S, Li Z, Lai Y, et al. HER2 copy number of circulating tumour DNA functions as a biomarker to predict and monitor trastuzumab efficacy in advanced gastric cancer. Eur J Cancer. 2018;88:92–100.
Shoda K, Masuda K, Ichikawa D, Arita T, Miyakami Y, Watanabe M, Konishi H, Imoto I, Otsuji E. HER2 amplification detected in the circulating DNA of patients with gastric cancer: a retrospective pilot study. Gastric Cancer. 2015;18(4):698–710.
Aguilar-Mahecha A, Joseph S, Cavallone L, Buchanan M, Krzemien U, Batist G, Basik M. Precision medicine tools to guide therapy and monitor response to treatment in a HER-2+ gastric cancer patient: case report. Front Oncol. 2019;9:698.
Jogo T, Nakamura Y, Shitara K, Bando H, Yasui H, Esaki T, Terazawa T, Satoh T, Shinozaki E, Nishina T, et al. Circulating tumor DNA analysis detects FGFR2 amplification and concurrent genomic alterations associated with FGFR inhibitor efficacy in advanced gastric cancer. Clin Cancer Res. 2021;27(20):5619–27.
Wang DS, Liu ZX, Lu YX, Bao H, Wu X, Zeng ZL, Liu Z, Zhao Q, He CY, Lu JH, et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut. 2019;68(7):1152–61.
Wang Y, Zhao C, Chang L, Jia R, Liu R, Zhang Y, Gao X, Li J, Chen R, **a X, et al. Circulating tumor DNA analyses predict progressive disease and indicate trastuzumab-resistant mechanism in advanced gastric cancer. EBioMedicine. 2019;43:261–9.
Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, Lauwers GY, Christensen JG, Wilner KD, Haber DA, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29(36):4803–10.
Lee J, Kim ST, Kim K, Lee H, Kozarewa I, Mortimer PGS, Odegaard JI, Harrington EA, Lee J, Lee T, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: the VIKTORY umbrella trial. Cancer Discov. 2019;9(10):1388–405.
Du J, Wu X, Tong X, Wang X, Wei J, Yang Y, Chang Z, Mao Y, Shao YW, Liu B. Circulating tumor DNA profiling by next generation sequencing reveals heterogeneity of crizotinib resistance mechanisms in a gastric cancer patient with MET amplification. Oncotarget. 2017;8(16):26281–7.
Frigault MM, Markovets A, Nuttall B, Kim KM, Park SH, Gangolli EA, Mortimer PGS, Hollingsworth SJ, Hong JY, Kim K, et al. Mechanisms of acquired resistance to savolitinib, a selective MET inhibitor in MET-amplified gastric cancer. JCO Precis Oncol. 2020;4:222–32.
Distler J, Tetzner R, Weiss G, König T, Schlegel A, Bagrowski M. Evaluation of different blood collection tubes and blood storage conditions for the preservation and stability of cell-free circulating DNA for the analysis of the methylated (m)SEPT9 colorectal cancer screening marker. Adv Exp Med Biol. 2016;924:175–8.
Warton K, Lin V, Navin T, Armstrong NJ, Kaplan W, Ying K, Gloss B, Mangs H, Nair SS, Hacker NF, et al. Methylation-capture and next-generation sequencing of free circulating DNA from human plasma. BMC Genomics. 2014;15(1):476.
Acknowledgements
Not applicable.
Funding
This study was supported by Shanghai Commission of Science and Technology (20Y11908100).
Author information
Authors and Affiliations
Contributions
JHL and DYZ wrote the original draft of the manuscript. JMZ and LD critically reviewed, edited, and approved the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, JH., Zhang, DY., Zhu, JM. et al. Clinical applications and perspectives of circulating tumor DNA in gastric cancer. Cancer Cell Int 24, 13 (2024). https://doi.org/10.1186/s12935-024-03209-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03209-4