
Abstract
Background
The sesquiterpene germacrene A is a direct precursor of ß-elemene that is a major component of the Chinese medicinal herb Curcuma wenyu** with prominent antitumor activity. The microbial platform for germacrene A production was previously established in Saccharomyces cerevisiae using the germacrene A synthase (LTC2) of Lactuca sativa.
Results
We evaluated the performance of LTC2 (LsGAS) as well as nine other identified or putative germacrene A synthases from different sources for the production of germacrene A. AvGAS, a synthase of Anabaena variabilis, was found to be the most efficient in germacrene A production in yeast. AvGAS expression alone in S. cerevisiae CEN.PK2-1D already resulted in a substantial production of germacrene A while LTC2 expression did not. Further metabolic engineering the yeast using known strategies including overexpression of tHMGR1 and repression of squalene synthesis pathway led to an 11-fold increase in germacrene A production. Site-directed mutagenesis of AvGAS revealed that while changes of several residues located within the active site cavity severely compromised germacrene A production, substitution of Phe23 located on the lateral surface with tryptophan or valine led to a 35.2% and 21.8% increase in germacrene A production, respectively. Finally, the highest production titer of germacrene A reached 309.8 mg/L in shake-flask batch culture.
Conclusions
Our study highlights the potential of applying bacterial sesquiterpene synthases with improved performance by mutagenesis engineering in producing germacrene A.
Similar content being viewed by others


Introduction
Terpenoids (terpenes/isoprenoids) are the most abundant and the largest class of natural products that are widely used as pharmaceuticals, herbicides, flavorings, fragrances, and biofuels [1, 2]. Natural extraction of trace and valuable terpenoids from their native sources, mostly plants, requires large amounts of plant materials, tedious procedures and high production cost, which is far from satisfying their market demand. With development of metabolic engineering and synthetic biology, platform microorganisms have been employed as cell factories for efficient synthesis of terpenoids. The budding yeast Saccharomyces cerevisiae is one particularly promising host for terpenoid production due to its generally recognized status as safe and robustness in fermentation. Specifically, S. cerevisiae has a native mevalonate (MVA) pathway that can be exploited to effectively supply isoprenoid precursors.
Several metabolic engineering strategies have been well established to increase the yield of terpenoid products in S. cerevisiae [3,4,5,6]. One critical step is to increase the precursor supply, which can be achieved by overexpressing a truncated version of the rate-limiting HMG-CoA reductase 1 (tHMGR1) [7], the farnesyl pyrophosphate (FPP) synthase ERG20 [8], and the transcription factor UPC2-1 [9, 10]. Together these strategies promote flux through the MVA pathway in cases of synthesizing all classes of terpenes. Moreover, downregulation of the competing squalene synthesis pathway by replacing the native ERG9 promoter with repressive promoters like MET3 [8] or HXT1 [11], is another effective strategy to increase the supply of FPP in mono-, di- and sesquiterpenes production. In addition to boosting precursor supply, highly efficient terpene synthases represent a crucial “pulling” step for biosynthesis of target products with high yield. To this end, strategies such as screening efficient synthases from various sources, enhancing enzyme expression via codon optimization, improving enzyme properties via protein engineering, and expressing fused enzymes catalyzing adjacent reactions, have been employed.
Terpenoid synthases are responsible for the cyclization and/or rearrangement of linear isoprenoid diphosphates into cyclic terpenoids [12, 13]. Given the vast variety of terpenoids, it is anticipated that the exact architecture of the active site cavity is strictly defined in specific synthases [13]. This is particularly true in the case of sesquiterpene synthases that catalyze reactions to yield a myriad of structurally diverse C15-hydrocarbons using the same substrate FPP [14]. Due to the complexity of the catalytic reaction cascade, rational engineering of terpenoid synthases to change product specificity or to enhance activity remains a challenge [12, 15]. However, sequence comparisons among closely related but functionally different enzymes combined with mutagenesis to change synthase performance have been proved to be effective in quite a few cases [12, 16, 17].
Sesquiterpene ß-elemene, a major component in Chinese medicinal herb, Curcuma wenyu** T. H. Chen et C. Ling, displays antitumor activity against a variety of tumor types [18] and has been clinically administrated to tumor treatments. The sesquiterpene synthase for ß-elemene has not been identified from Curcuma wenyu** and other related plants. Instead, it is generally considered that ß-elemene is transformed from germacrene A, which is synthesized by germacrene A synthase (GAS). Under heating or acidic conditions, germacrene A is automatically converted to ß-elemene via one-step intramolecular rearrangement. The in vitro transformation occurs even at room temperature [19]. Therefore, synthesis of germacrene A by dedicated synthases is a promising approach to efficiently produce ß-elemene, which would greatly lower its cost of production. Hu et al. have successfully established a production platform to produce germacrene A by expressing a GAS (LTC2) of Lactuca sativa in a yeast chassis strain that has been optimized with isoprenoid production [20]. Germacrene A production was further increased by introduction of one more copy of tHMGR1 expression cassette and expression of fused LTC2 and ERG20 with a final titer of 190.7 mg/L in shake flasks [20]. Moreover, Zhang et al. [21] have reported the development of coupling techniques for thermal conversion of germacrene A to ß-elemene, which reduces the production cost of β-elemene to 0.15% of that from plant extraction.
In this study, we aimed to improve germacrene A biosynthesis in engineered S. cerevisiae (Fig. 1). We first performed screening with several identified or putative GASs from different sources, and found that a cyanobacterial synthase, AvGAS, showed the best performance on germacrene A synthesis. Known strategies of engineering the MVA pathway including expression of tHMGR1 using a constitutive promoter and repression of squalene synthesis pathway via replacing the EGR9 promoter with the HXT1 promoter, further improved germacrene A production. Site-directed mutagenesis of AvGAS was simultaneously performed and AvGAS F23W mutant was found to enable recombinant yeast strains to produce 309.8 mg/L germacrene A in shake-flask cultivation.

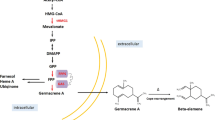
Schematic overview of germacrene A production in S. cerevisiae. To improve germacrene A production, GAS (marked in red) screening and site-directed mutagenesis (represented with an asterisk) were performed, the tHMGR1 gene (marked in purple) was overexpressed, the LPP1 and DPP1 genes (marked in green) were deleted, and the ERG9 gene (marked in blue) was downregulated by replacing its native promoter with the promoter of HXT1. Acetyl-CoA: acetyl coenzyme A, HMG-CoA: 3-hydroxy-3-methylglutaryl-CoA, tHMGR1: truncated HMG-CoA reductase gene, IPP: isopentenyl pyrophosphate, DMAPP: dimethylallyl pyrophosphate, ERG20: farnesyl diphosphate synthase gene, FPP: farnesyl diphosphate, FOH: farnesol, ERG9: squalene synthase gene, GAS: germacrene A synthase. Solid arrows represent one-step transformation while dashed arrows represent transformation comprising multiple intermediate steps
Results
Screening GASs with better performance on germacrene A synthesis
GAS catalyzes the reaction to synthesize germacrene A from FPP. To increase germacrene A production, several identified and putative GASs from different species were expressed in S. cerevisiae (Fig. 2). Germacrene A synthesis in these recombinant strains wherein FPP was solely derived from the endogenous MVA pathway, was evaluated. Since germacrene A was completely converted to ß-elemene at high temperature during gas chromatography analysis, ß-elemene was applied as a standard for detection and quantification of germacrene A extracted from yeast cultures (Additional file 1: Figure S1). Preliminary screening was performed with three GASs previously identified from plants including Achillea millefolium [22], Taraxacum officinale [23] and Lactuca sativa [20], and one identified from the bacterium Nostoc sp. PCC 7120 [24]. As shown in Fig. 3, expression of the three plant-derived GASs in S. cerevisiae led to no detectable germacrene A production compared with the parent S. cerevisiae CEN.PK2-1D strain. In contrast, expression of the cyanobacterial GAS (NsGAS) resulted in a significant production of germacrene A. We therefore chose this synthase for the next round of screening.

Phylogenetic analyses of the putative GASs used for enzyme screening and their orthologues. Previously known GASs were indicated by triangles. AvGAS identified in this study with the best performance on germacrene A production was indicated with an asterisk. The ten synthases applied for screening in this study were colored orange. The protein sequences of putative GASs or their orthologues were retrieved from NCBI or Uniprot databases with accession or entry numbers as follows: NsGAS (NCBI accession NO. WP_010998816), AmGAS (NCBI accession NO. AGD80135), ToGAS (NCBI accession NO. ALY05868), LsGAS (LTC2, NCBI accession NO. AAM11627), AvGAS (UniProt entry NO. Q3MBN2), NcGAS (UniProt entry NO. A0A1Z4HUB4), MmGAS (UniProt entry NO. V5BIL8), CsGAS (UniProt entry NO. A0A1Z4H7M3), SpGAS (UniProt entry NO. A0A1Y2N8F8), PsGAS (UniProt entry NO. A0A171DER6), NlGAS (NCBI accession NO. WP_190654468), MlGAS (NCBI accession NO. WP_024299200), SgGAS (NCBI accession NO. WP_190143472), SeGAS (NCBI accession NO. WP_060893906), PpGAS (NCBI accession NO. WP_189237389), NgPS (Pentalenene synthase, NCBI accession NO. SBO99268)

Screening GASs with better performance on germacrene A synthesis. Expression of the synthases from various sources were performed in the parent S. cerevisiae CEN.PK2-1D strain. Values represent the means of three biological replicates. Error bars are the standard deviations from these replicates
Protein blast was performed in UniProt database with NsGAS as a query and bacterial orthologues sharing various sequence identities with NsGAS were retrieved. Six candidates were selected for further analysis, including putative GASs of Anabaena variabilis ATCC29413 (AvGAS, identity 93.2%), Nostoc carneum NIES-2107 (NcGAS, identity 70.8%), Calothrix sp. NIES-2100 (CsGAS, identity 55%), Methyloglobulus morosus KoM1 (MmGAS, identity 38.6%), Streptomyces platensis (SpGAS, identity 31%) and Planomonospora sphaerica (PsGAS, identity 27%) (Fig. 2; Additional file 1: Figure S2). Only two synthases, NcGAS and AvGAS, conferred germacrene A-producing capability on yeast cells, indicating that these two synthases are effective GASs. While NcGAS expression led to much less ß-elemene synthesis than NsGAS, expression of AvGAS resulted in a twofold increase in ß-elemene production reaching a titer of 17.8 mg/L in the corresponding yeast strain SGAS5. These results indicate that AvGAS of A. variabilis ATCC29413 exhibited the best performance on germacrene A production among the screened synthases when expressed in yeast, and was therefore selected for further engineering study.
Engineering the MVA pathway to improve germacrene A biosynthesis
FPP from the yeast MVA pathway is the direct precursor for sesquiterpene synthesis. To improve sesquiterpene production, it is necessary to enhance FPP availability. Frequently used strategies to metabolically engineer yeast chassis to increase FPP supply include overexpression of the rate-limiting enzyme tHMGR1 of the MVA pathway and repression of the competing squalene synthesis pathway and farnesol synthesis pathway [3,4,5,6]. Overexpression of tHMGR1 in strain SGAS5 resulted in strain SMVA1, which showed a 4.4-fold increase in germacrene A production, reaching a titer of 77.8 mg/L (Fig. 4). Repressing squalene synthesis in strain SMVA1 by replacing the ERG9 promoter with the HXT1 promoter [25, 26] was found to further increase germacrene A production up to 186.2 mg/L in the resultant SMVA2 strain (Fig. 4). Along with the enhanced germacrene A production, farnesol synthesis was also significantly increased due to the enhanced FPP supply (Additional file 1: Figure S3). Two phosphatase-encoding genes, DPP1 and LPP1, involved in farnesol synthesis were therefore deleted in strain SMVA2 to obtain the SMVA3 strain. Whereas farnesol accumulation was decreased by 24.6%, germacrene A production in SMVA3 was not significantly increased (Fig. 4; Additional file 1: Figure S3).

Germacrene A production was enhanced by increasing the precursor FPP availability in yeast. Strategies include overexpression of tHMGR1, repression of ERG9 expression by replacing its native promoter with HXT1 promoter, and deletion of DPP1 and LPP1. Values represent the means of three biological replicates. Error bars are the standard deviations from these replicates
Engineering AvGAS for improved germacrene A biosynthesis via site-directed mutagenesis
While enhanced FPP supply resulted in not only increased production of the target product, but also accumulation of the byproduct farnesol, implying that FPP flux towards germacrene A synthesis is becoming a limiting step. We therefore attempted to engineer AvGAS to improve its catalytic capability of synthesizing germacrene A via site-directed mutagenesis. Homology modelling was first performed with the resolved structure of a bacterial 1,8-cineole synthase in complex with a FPP analogue (PDB ID: 5NX6) [14] as a template. As shown in Fig. 5a, like most bacterial terpene synthases, modelled AvGAS adopts a common α-helical fold with a single catalytic domain, but lacks the additional N-terminal α-barrel domain characteristic of plant enzymes [27]. Two conserved regions within the active site, the aspartate-rich (DDXX(X)(D,E)) motif and the NSE (NDXXSXX(R,K)(E,D)) triad [14, 28], required for binding catalytically essential Mg2+ ions, were also present in AvGAS (Additional file 1: Figure S4).

Modelled structure of AvGAS (a) and molecular docking simulation with substrate FPP (b). FPP is represented as cyan sticks and amino acids as indicted are shown as green sticks
Site-directed mutagenesis of residues located in the catalytic pocket of AvGAS severely compromised germacrene A biosynthesis
Based on the modelled structure of AvGAS, molecular docking simulation of AvGAS with substrate FPP was performed (Fig. 5b). In addition to coordination by Mg2+ ions, the pyrophosphate moiety of FPP makes interactions with residues including Arg310, Asn222, and Tyr311 (Fig. 5b). The hydrophobic FPP carbon chain interacts with several residues including Leu54, Leu78, Ala181, and Ile 215 within the substrate binding pocket via Van der Waals' force. While residues involved in binding the pyrophosphate moiety of FPP are evolutionarily conserved in terpene synthases, it is the hydrophobic cavity essentially mediates the sequential FPP carbon chain cyclization [14], We therefore attempted to perform mutations on residues Leu78 and Ala181 as well as residues Leu55, Ala182, Ala183, and Ile75 that are also located within the substrate binding cavity. Strain SHEF was first constructed with CEN.PK2-1D as the parent strain for evaluation of various AvGAS site-directed mutants, wherein tHMGR1 was overexpressed, the EGR9 promoter was replaced with the HXT1 promoter, and LPP1 and DPP1 were deleted. The wild type (WT) and resultant mutants of AvGAS were expressed in the SHEF strain. Whereas substitution of Leu78 with Ala, Thr, Ile, Val, or Gly led to significantly decreased germacrene A production, its replacement with Phe completely abolished production of the target product (Fig. 6a). Similarly, mutations of Ala181 (to Cys, Gly, Phe, Leu, Tyr, or Thr), Ala182 (to Thr, Gly, Ser, Phe or Leu), Ala183 (to Phe, Ser, Thr, Gly or Leu), Leu55 (to Phe, Trp, Val, Ala or Tyr), and Ile75 (to Ala, Leu, Tyr or Phe) all resulted in markedly decreased or undetectable germacrene A accumulation (Fig. 6b–f). These results indicate that residues located in the catalytic cavity of AvGAS play key roles in biosynthesis of germacrene A.

Production of germacrene A of yeast strains expressing AvGAS mutants carrying site-directed mutagenesis of residues located within the active site cavity. Mutations were performed on residues including L78 (a), A181 (b), A182 (c), A183 (d), L55 (e), and I75 (f). Values represent the means of three biological replicates. Error bars are the standard deviations from these replicates
Expression of the AvGAS mutants F23W and F23V improved germacrene A biosynthesis
Since mutation of residues within the catalytic cavity of AvGAS failed to produce a mutant enzyme with improved capability in germacrene A biosynthesis, we next analyzed the impact of mutations of some “variable” residues whose change may contribute to enzyme activity. AvGAS and its two orthologues with relatively higher identities, NsGAS and NcGAS, are assumed to display significant different enzymatic activities since their expression in yeast led to different germacrene A accumulation as described above. Sequence alignment of AvGAS with NsGAS and NcGAS revealed that three residues located far from the active site cavity were identical at the corresponding positions for NsGAS and NcGAS (Val23, Tyr152, and Asn268), but distinct in AvGAS (Phe23, His152 and Lys268) (Fig. 7a). To see whether changes in these residues could have an effect on the enzymatic activity of AvGAS, Phe23 was replaced with Val, Tyr, Leu, and Trp, respectively, His152 was changed to Tyr, Ala, Gly, and Leu, respectively, and Lys268 was substituted with Arg, Asp, and Glu, respectively. Unlike mutations within the active site cavity, none of these mutations abolished germacrene A synthesis (Fig. 7b, c). Expression of a majority of these mutants allowed yeast cells to accumulate decreased or similar levels of germacrene A compared to cells expressing WT AvGAS. Notably, two yeast strains expressing AvGAS-F23W and AvGAS-F23V displayed markedly improved germacrene A production (35.2% and 21.8% increase, respectively). The production titer of germacrene A in strains SHEF-F23W and SHEF-F23V reached 309.8 mg/L and 278.9 mg/L, respectively. The increase in product accumulation is not due to improved cell growth since SHEF-F23W and SHEF-F23V showed similar biomass accumulation with other AvGAS mutant strains (Additional file 1: Table S1).

Production of germacrene A of yeast strains expressing AvGAS mutants carrying site-directed mutagenesis of residues Phe23, His152, and Lys268. a Sequence alignment of AvGAS with its two highly identical orthologues NsGAS and NcGAs. Phe23, His152, and Lys268 are indicated by blue triangles. b, c The effect of site-directed mutations of His152 (b), Phe23 and K268 (c) on germacrene A production-producing capability of AvGAS when expressed in yeast. Values represent the means of five biological replicates. Error bars are the standard deviations from these replicates
Discussion
The sesquiterpene germacrene A is a direct precursor of ß-elemene that exhibits therapeutic efficacy against various types of tumors [18]. In this study, we combined GAS screening, site-directed mutagenesis engineering, and metabolic engineering of yeast chassis to improve germacrene A production.
In a previous study to increase germacrene A production in yeast, Hu et al. [20] has evaluated the capability of two GASs from plants and selected LTC2 of Lactuca sativa to achieve a relatively high production yield. Here, in addition to LTC2 (here termed LsGAS), we also included nine more known or putative GASs from plants or bacteria for evaluation. When expressed in a S. cerevisiae strain without the MVA pathway engineering, only three cyanobacterial synthases, NsGAS, NcGAS, and AvGAS, showed capability of synthesizing germacrene A. The inability of LTC2 (LsGAS) to catalyze the detectable germacrene A formation in the present study may result from the use of yeast strain with a different genetic background. Hu et al. employed a parent strain with overexpression of tHMGR1 and ERG20, repression of ERG9, and deletion of DPP1 and LPP1, wherein the MVA flux and FPP supply was significantly enhanced. LTC2 (LsGAS) and TmGAS were indeed found to successfully mediate germacrene A production, respectively, in an engineered yeast strain with tHMGR1 overexpression and ERG9 repression (Additional file 1: Figure S5). Nonetheless, the detected target product synthesis was still markedly lower than those resultant from the respective expression of NsGAS, NcGAS, and AvGAS (Additional file 1: Figure S5), supporting that these three cyanobacterial synthases exhibited much better performance in germacrene A synthesis compared to LTC2 (LsGAS). Our result is consistent with the previous report that bacterial terpene synthase outperforms the orthologous plant enzyme: bLinS of Streptomyces clavuligerus produced about 300-fold more linalool than RLinS_Aa of Artemisia annua [14]. Structural analyses revealed that, unlike plant enzymes comprising the N-terminal α-barrel domain and the C-terminal α-helix domain, the majority of bacterial terpene sesquiterpene synthases contain only the α-helices constituting a single catalytic domain. In addition to higher catalytic activity, the smaller size and simplified structure of bacterial synthases might facilitate its expression in yeast and hence contribute to the catalyzed formation of target product. These possibilities definitely warrants further investigation.
Increasing precursor supply is crucial in ensuring high-level target product synthesis. When some routinely used strategies were applied to enhance precursor FPP supply in yeast, a marked increase in germacrene A production was observed. However, we noticed that the byproduct farnesol also increased with the target product. Although the two phosphatase encoding genes DPP1 and LPP1 were deleted, farnesol accumulation was not completely eliminated. It is highly probable that other as yet to be identified phosphatases exist to compensate for the loss of DPP1 and LPP1, such as PHO8, APP1, and PAH1 [29, 30]. Despite the increased farnesol production, Hu et al. reported that introduction of one additional copy of tHMGR1 expression cassette led to a further 1.5-fold increase in germacrene A production, indicating that directing more flux into the MVA pathway to enhance the precursor pool is beneficial for germacrene A accumulation. We also noticed that farnesol production in yeast strain SHEF expressing AvGAS is markedly decreased compared to that expressing plant LsGAS, and a further reduction was observed in SHEF strain expressing AvGAS F23W mutant, suggesting that efficient GAS contribute to shifting the flux from farnesol synthesis to germacrene A synthesis (Additional file 1: Figure S6). Furthermore, it was observed that germacrene A was also trapped intracellularly, when we broke SHEF cells, and found that germacrene A accumulates in intracellular fractions (Additional file 1: Figure S7). Future studies on identification of the membrane efflux transporters which could efficiently export germacrene A to culture supernatant might contribute to decreasing intracellular accumulation and therefore enhancing final extracellular yield.
Compared to the various already known strategies to effectively engineering the S. cerevisiae MAV pathway, terpene synthases responsible for the cyclization of linear isoprenoid diphosphates into cyclic terpenoids are intractable to be engineered [12, 15]. The difficulty mainly lies in the complexity of the reaction cascade occurring within the catalytic cavity of the synthase and the lack of high-throughput screening method. Indeed, repeated attempts to target residues within the active site cavity of AvGAS to improve its activity failed, as demonstrated by the undetectable or markedly decreased germacrene A production with the relevant AvGAS mutants (Fig. 6). It is thus highly probable that most, if not all, residues along the cavity may directly or indirectly participate in the sequential cyclization of FPP and thus play important or essential roles in the catalytic process. Given that the same substrate FPP can be transformed into a myriad of various sesquiterpenes by dedicated synthases, one can speculate that the exact shape and size as well as the hydrophobicity of the active site cavity of the respective synthase is vitally essential for specific product formation. The observation that variation of Ala181 to Gly with a similar size decreased but not abolished germacrene A production, whereas its replacement with larger residues including Cys, Phe, Leu, Tyr or Thr resulted in undetectable production of germacrene A, suggests that maintenance of the precise configuration of the active site cavity is crucial for the efficient carryon of the reaction cascade. Similar phenomenon was also observed for Ala182 and Ile75 wherein mutation of Ala to Gly and Ile to Leu resulted in the mildest impact on germacrene A yield among all the tested mutations. However, exceptions to the above scenario also exist. Variation of Leu78 to Ile with similar size and hydrophobicity resulted in a more severe impact on germacrene A production compared to replacement by Ala with smaller size and lower hydrophobicity. Moreover, while A183G was more efficient in product synthesis than A183F, change of Ala to hydrophilic Ser (A183S) outperformed both A183G and A183F. This discrepancy further implicates that any subtle perturbation of the precise configuration of the active site cavity or the intricate interaction network between enzyme and substrate might exert a significant disturbing effect on the enzyme activity. Possibility also can not be excluded that the relevant mutations just destroy the overall structure of AvGAS and thus reduced the solubility of the expressed synthase in yeast cells. Of the three cyanobacterial germacrene A synthases, AvGAS showed the best performance in germacrene A biosynthesis when expressed in yeast. Whereas the reason for the higher capability of AvGAS as compared to the other two highly identical orthologues is not clear at present, sequence alignment did reveal various residues in AvGAS that are distinct from its corresponding orthologues. Three such residues including His152, Phe23, and Lys268 were selected for site-directed mutagenesis. Whereas quite a few mutants did not affect germacrene production, two resultant mutants AvGAS-F23W and AvGAS-F23V enabled improved germacrene A production compared with WT AvGAS, resulting in an increase of 35.2% and 21.8% in production yield, respectively. In contrast, mutants H152A, H152L, and H152G exhibited decreased capability in germacrene A production. These substitutions most probably affected the hydrophilicity of His152 site and thus exerted a negative effect on the performance of AvGAS. Considering that Phe23 is located on the lateral surface of AvGAS where is relatively far way from the active site cavity (Additional file 1: Figure S8), it is speculated that these two mutations exert their effect via interacting with its proximal residues which then indirectly affects substrate accommodation or product release and thus germacrene A synthesis. Possibility can not be excluded that these two beneficial mutations of Phe23 facilitated functional AvGAS soluble expression in yeast cells, which contributes to the observed increase in product yield. It can be expected that more strategies such as introduction of one more copy of tHMGR1, overexpression of ERG20, and appropriate fusion of ERG20 with the cyanobacterial synthase, which were employed in previous biosynthesis of germacrene A [20], would further increase the yield to a large margin.
Materials
Strains and culture conditions
Escherichia coli DH5α was used for routine plasmid construction and was cultivated at 37 °C in Luria–Bertani (LB) medium. When necessary, 100 µg/mL of ampicillin was supplemented in LB medium. Parent strain S. cerevisiae CEN.PK2-1D was cultured at 28 °C in yeast extract peptone dextrose (YPD) medium. Engineered yeast strains were cultivated in synthetic complete (SC) drop-out media, and 50 µg/mL of leucine, histidine, tryptophan, uracil, or 40 µg/mL of geneticin (G418) were individually or simultaneously added when necessary.
Plasmids and strains construction
All the plasmids and strains used in this study were listed in Table 1. For GAS screening, totally ten identified or putative GAS encoding genes or cDNA sequences including NsGAS (NCBI accession NO. WP_010998816), AmGAS (NCBI accession NO. AGD80135), ToGAS (NCBI accession NO. ALY05868), LsGAS (NCBI accession NO. AAM11627), AvGAS (UniProt entry NO. Q3MBN2), NcGAS (UniProt entry NO. A0A1Z4HUB4), MmGAS (UniProt entry NO. V5BIL8), CsGAS (UniProt entry NO. A0A1Z4H7M3), SpGAS (UniProt entry NO. A0A1Y2N8F8), and PsGAS (UniProt entry NO. A0A171DER6), were codon-optimized according to the S. cerevisiae codon bias and synthesized by GenScript Biotech Corporation (Suzhou, China). These GAS gene fragments were ligated into the pRS425 plasmid carrying the GAL1 promoter and the ADH1 terminator that were both amplified from S. cerevisiae W303 genomic DNA. The resultant plasmids for expression of GASs were individually transformed into S. cerevisiae CEN.PK2-1D using the PEG/LiAc method [31]. To overexpress tHMGR1, a truncated version of HMGR1 encoding fragment [7] was amplified with S. cerevisiae CEN.PK2-1D genomic DNAas template, and ligated into the pRS306 plasmid that harbors the TPI1 promoter and the CYC1 terminator. The resultant plasmid pRS306-tHMGR1 was transformed into strain SGAS5 to generate SMVA1. The HXT1 promoter and the loxp-KanMX-loxp cassette were amplified from S. cerevisiae CEN.PK2-1D genomic DNA and plasmid pUG6 [32], respectively, and fused together via overlap extension PCR [33]. A pair of primers containing 70-bp nucleotide fragments upstream and downstream of the native ERG9 promoter, respectively, were used to amplify the fused fragment, to construct the ERG9 promoter replacement cassette. This cassette was subsequently integrated into strain SMVA1, resulting in strain SMVA2 wherein the native ERG9 promoter was replaced by the HXT1 promoter. To delete DPP1, a pair of primers containing 70-bp nucleotide fragments upstream and downstream of the DPP1 gene was used to amplify the loxp-KanMX-loxp cassette to construct DPP1 gene deletion cassette. This cassette was integrated into SMVA2 wherein the KanMX selection marker was removed ahead of time. The deletion cassette of LPP1 was constructed similarly, and integrated into strain SMVA2 with deleted DPP1, resulting in strain SMVA3 wherein DPP1 and LPP1 were both deleted. To construct strain SHEF, pRS306-tHMGR1, ERG9 promoter replacement cassette, and DPP1 and LPP1 deletion cassettes were integrated into S. cerevisiae CEN.PK2-1D in succession using the same procedures as described above. To generate AvGAS fragments with specific site-directed mutations, overlap-extension PCR was performed with pRS425-AvGAS as template. The mutated fragments were ligated into the pRS425 plasmid carrying the GAL1 promoter and the ADH1 terminator. DNA sequencing was performed to confirm that each mutagenesis occurred as expected. The pRS425-derived plasmids containing the expression cassette of WT and various site-specific mutated AvGAS genes were transformed into strain SHEF, respectively.
Homology modelling
A high resolution protein structure with significant homology to the AvGAS protein sequence was obtained from the Protein Data Bank (PDB) using NCBI BLAST. This structure (PDB ID: 5NX6) was used as a template for subsequent homology modelling which was performed with the Molecular Operating Environment (MOE) 2014.09 software package. After removing uncorrelated ligands, target and template sequences were compared to define conserved regions. Ten transition models were obtained by permutation and combination of candidate loop regions as well as side-chain rotamers. These ten candidates were then subjected to energy optimization with the AMBER12/EHT force field and R-field implicit solvent. The quality of each candidate was evaluated using the GB/VI scale. The highest-scoring model was chosenfor further energy optimized.
Molecular docking
The docking module Dock in the MOE software was used to predict the binding between AvGAS and the substrate FPP. The 2D structure of FPP was drawn with ChemBioDraw 2014, and a 3D structure was obtained with MOE by energy optimization. The protonation state of proteins and the location of hydrogen atoms was obtained using the LigX module in MOE, at pH 7 and 300 K. Before molecular docking, the AMBER12:EHT force field and the R-field implicit solvent model were selected. Docking was completed in the induced fit mode; the side chains in the binding pocket were automatically adjusted according to ligand conformation. The weight that restrained the rotation of the side chains was set at 10. The binding modes of FPP were first ranked by a London dG scoring function. The first 30 conformations were re-assessed following the GBVI/WSA dG method. The interactions between FPP and proteins were graphically represented using PyMOL (www.pymol.org).
Analysis of germacrene A production by engineered yeast strains
Recombinant yeast transformants were inoculated into 5 mL of SC liquid media containing 1% glucose as carbon source. After 24 h growth, equal amount of yeast cells were collected and transferred to 30 mL PY (2% peptone plus 1% yeast extract) liquid media containing 0.1% glucose and 2% galactose, with initial OD600 of 0.5 measured by a microplate reader (BioTek, USA). All flasks were immediately supplemented with 20% (vol/vol) dodecane after seeding. After 72-h cultivation, 100 μL of cell culture was taken and diluted for measuring OD600 using the microplate reader (BioTek, USA). The rest was centrifuged at 13,000 rpm for 20 min, and 0.5 mL of the upper dodecane layer were collected and stored at -20 °C. To analyze whether germacrene A was trapped intracellularly, 30 mL of SHEF yeast cells were collected, washed with 25 mM phosphate buffer at pH 7.5, and resuspended in the same buffer containing 25 U lyticase. After treatment at 30 °C for 30 min, cells were collected and resuspended, followed by disruption using glass beads in a bead beater with 6000 rpm for 15 s, for three times. Cell lysates were subject to centrifugation at 8000 rpm for 10 min, and the supernatant was separated, with an addition of 50% dodecane (vol/vol). The mixture was vortexed for 10 min, and then centrifuged at 13,000 rpm for 20 min. The final upper dodecane layer were collected and stored. ß-elemene were identified using a GC–MS system (Shimadzu Co., Kyoto, Japan) equipped with an RTX-1 column (30 m × 0.25 mm × 0.25 μm). One microliter of each dodecane sample was injected into the system with a split ratio of 10 and the carrier gas helium was set at a constant flow rate of 0.78 mL/min. The oven temperature was first maintained at 40 °C for 2 min, and then gradually increased to 160 °C at a rate of 10 °C /min, held for 2 min, and finally increased to 250 °C at a rate of 15C /min and held for 5 min. The mass spectrometer was set to the SIM acquisition mode, scanning m/z ions within the range 50–650 for identification of ß-elemene. For quantification of ß-elemene, a GC-FID system (Shimadzu Co., Kyoto, Japan) equipped with an RTX-1701 column (30 m × 0.25 mm × 0.25 μm) was applied. One microliter of each dodecane sample was injected into the system with a split ratio of 10 and the carrier gas nitrogen was set at a constant flow rate of 2.34 mL/min. The oven temperature was first maintained at 40 °C for 1 min, and then gradually increased to 180 °C at a rate of 25 °C /min, held for 3 min, and finally increased to 250 °C at a rate of 15 °C/min and held for 5 min. The total run time was 20 min. ß-elemene standard was dissolved in dodecane and used to plot standard curves for quantification. Germacrene A production are presented as ß-elemene equivalents.
Sequence analysis and phylogenetic tree construction
The amino acid sequences of GASs were retrieved from NCBI or Uniprot databases. Sequence alignments were performed using Clustal W [34]. The phylogenetic tree was constructed using the neighbour-joining method with MEGA7.0 [35]. Numbers on the tree branches represent the bootstrap support calculated per 1000 bootstrap replicates.
Conclusions
By combining GAS screening, enzyme mutagenesis engineering, and metabolic engineering, production of germacrene A in yeast was markedly improved. GAS enzyme screening showed that three cyanobacterial GASs, especially AvGAS, outperformed plant orthologues in germacrene A production in yeast. Simultaneously metabolic engineering of yeast strain to enhance the MVA pathway flux and FPP supply dramatically improved germacrene A production by 11-fold. Finally, site-directed mutation of AvGAS Phe23 to tryptophan or valine led to a further 35.2% and 21.8% increase in germacrene A accumulation. These results highlight the potential of bacterial terpene synthases in sesquiterpene production in engineered yeast and provide insights into the mutagenesis engineering of sesquiterpene synthases.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- GAS:
-
Germacrene A synthase
- MVA:
-
Mevalonate
- tHMGR1:
-
Truncated HMG-CoA reductase 1
- FPP:
-
Farnesyl pyrophosphate
- LB:
-
Luria–Bertani
- YPD:
-
Yeast extract peptone dextrose
- SC:
-
Synthetic complete
- PDB:
-
Protein Data Bank
- MOE:
-
Molecular Operating Environment
- GC:
-
Gas chromatography
- MS:
-
Mass spectroscopy
References
Tholl D. Biosynthesis and biological functions of terpenoids in plants. Adv Biochem Eng Biotechnol. 2015;148:63–106.
Zhang C, Hong K. Production of terpenoids by synthetic biology approaches. Front Bioeng Biotechnol. 2020;8:347.
Zhang Y, Nielsen J, Liu Z. Engineering yeast metabolism for production of terpenoids for use as perfume ingredients, pharmaceuticals and biofuels. FEMS Yeast Res. 2017;17:fox080.
Zhuang X, Chappell J. Building terpene production platforms in yeast. Biotechnol Bioeng. 2015;112:1854–64.
Paramasivan K, Mutturi S. Progress in terpene synthesis strategies through engineering of Saccharomyces cerevisiae. Crit Rev Biotechnol. 2017;37:974–89.
Vickers CE, Williams TC, Peng B, Cherry J. Recent advances in synthetic biology for engineering isoprenoid production in yeast. Curr Opin Chem Biol. 2017;40:47–56.
Polakowski T, Stahl U, Lang C. Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl Microbiol Biotechnol. 1998;49:66–71.
Westfall PJ, Pitera DJ, Lenihan JR, Eng D, Woolard FX, Regentin R, Horning T, Tsuruta H, Melis DJ, Owens A, et al. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci USA. 2012;109:E111-118.
Crowley JH, Leak FW Jr, Shianna KV, Tove S, Parks LW. A mutation in a purported regulatory gene affects control of sterol uptake in Saccharomyces cerevisiae. J Bacteriol. 1998;180:4177–83.
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–3.
Scalcinati G, Knuf C, Partow S, Chen Y, Maury J, Schalk M, Daviet L, Nielsen J, Siewers V. Dynamic control of gene expression in Saccharomyces cerevisiae engineered for the production of plant sesquitepene alpha-santalene in a fed-batch mode. Metab Eng. 2012;14:91–103.
Gao Y, Honzatko RB, Peters RJ. Terpenoid synthase structures: a so far incomplete view of complex catalysis. Nat Prod Rep. 2012;29:1153–75.
Karunanithi PS, Zerbe P. Terpene synthases as metabolic gatekeepers in the evolution of plant terpenoid chemical diversity. Front Plant Sci. 2019;10:1166.
Karuppiah V, Ranaghan KE, Leferink NGH, Johannissen LO, Shanmugam M, Ni Cheallaigh A, Bennett NJ, Kearsey LJ, Takano E, Gardiner JM, et al. Structural basis of catalysis in the bacterial monoterpene synthases linalool synthase and 1,8-cineole synthase. ACS Catal. 2017;7:6268–82.
Lauchli R, Rabe KS, Kalbarczyk KZ, Tata A, Heel T, Kitto RZ, Arnold FH. High-throughput screening for terpene-synthase-cyclization activity and directed evolution of a terpene synthase. Angew Chem Int Ed Engl. 2013;52:5571–4.
Degenhardt J, Kollner TG, Gershenzon J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry. 2009;70:1621–37.
Kushiro T, Shibuya M, Masuda K, Ebizuka Y. Mutational studies on triterpene syntheses: engineering lupeol synthase into beta-amyrin synthase. J Am Chem Soc. 2000;122:6816–24.
Lu JJ, Dang YY, Huang M, Xu WS, Chen XP, Wang YT. Anti-cancer properties of terpenoids isolated from Rhizoma Curcumae—a review. J Ethnopharmacol. 2012;143:406–11.
de Kraker JW, Franssen MCR, de Groot A, Konig WA, Bouwmeester HJ. (+)-Germacrene A biosynthesis—the committed step in the biosynthesis of bitter sesquiterpene lactones in chicory. Plant Physiol. 1998;117:1381–92.
Hu Y, Zhou YJ, Bao J, Huang L, Nielsen J, Krivoruchko A. Metabolic engineering of Saccharomyces cerevisiae for production of germacrene A, a precursor of beta-elemene. J Ind Microbiol Biotechnol. 2017;44:1065–72.
Zhang XL HL, Dai ZB, Wang D, Zhang LL, Guo J, Liu Y. Recombinant yeast and thereof. China; 2018.
Pazouki L, Memari HR, Kannaste A, Bichele R, Niinemets U. Germacrene A synthase in yarrow (Achillea millefolium) is an enzyme with mixed substrate specificity: gene cloning, functional characterization and expression analysis. Front Plant Sci. 2015;6:111.
Huber M, Ep** J, Schulze Gronover C, Fricke J, Aziz Z, Brillatz T, Swyers M, Kollner TG, Vogel H, Hammerbacher A, et al. A latex metabolite benefits plant fitness under root herbivore attack. PLoS Biol. 2016;14:e1002332.
Agger SA, Lopez-Gallego F, Hoye TR, Schmidt-Dannert C. Identification of sesquiterpene synthases from Nostoc punctiforme PCC 73102 and Nostoc sp. strain PCC 7120. J Bacteriol. 2008;190:6084–96.
Souza AA, Miranda MN, da Silva SF, Bozaquel-Morais B, Masuda CA, Ghislain M, Montero-Lomeli M. Expression of the glucose transporter HXT1 involves the Ser-Thr protein phosphatase Sit4 in Saccharomyces cerevisiae. FEMS Yeast Res. 2012;12:907–17.
Garcia-Albornoz M, Holman SW, Antonisse T, Daran-Lapujade P, Teusink B, Beynon RJ, Hubbard SJ. A proteome-integrated, carbon source dependent genetic regulatory network in Saccharomyces cerevisiae. Mol Omics. 2020;16:59–72.
Chen F, Tholl D, Bohlmann J, Pichersky E. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011;66:212–29.
Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–20.
Song L. A soluble form of phosphatase in Saccharomyces cerevisiae capable of converting farnesyl diphosphate into E E-farnesol. Appl Biochem Biotechnol. 2006;128:149–58.
Chae M, Han GS, Carman GM. The Saccharomyces cerevisiae actin patch protein App1p is a phosphatidate phosphatase enzyme. J Biol Chem. 2012;287:40186–96.
Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425.
Guldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–24.
Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2:924–32.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Acknowledgements
We thank Prof. Hou ** from Shandong University for kindly providing the Saccharomyces cerevisiae CEN.PK2-1D strain.
Funding
This work is supported by the grant from the National Key Research and Development Program of China (2019YFA0905700).
Author information
Authors and Affiliations
Contributions
WZ and WL designed this project. WZ, JG and ZW performed the experiments. All the authors analyzed the data. WZ and WL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Identification of germacrene A by GC-MS analysis. (A, B) GC analyses of the ß-elemene standard (A) and the culture extract of the yeast strain SGAS5. (C) GC-MS spectra of the chromatographic peak corresponding to ß-elemene in (B). Figure S2. List of pairwise protein sequence identity (%) comparison between ten GAS candidates applied for screening in this study. Figure S3. Extracellular farnesol production of yeast strains SGAS5, SMVA1, SMVA2, and SMVA3. Figure S4. Sequence alignment of AvGAS with NsGAS, NcGAS, and bLinS from Streptomyces clavuligerus (5NX6). Two conserved regions within the active site, the aspartate-rich (DDXX(X)(D,E)) motif and the NSE (NDXXSXX(R,K)(E,D)) triad, required for binding catalytically essential Mg2+ ions, were indicated with boxes. Figure S5. Extracellular germacrene A production of yeast cells expressing the indicated GASs, respectively. GASs were expressed in S. cerevisiae CEN.PK2-1D wherein tHMGR1 was overexpressed and ERG9 was downregulated via promoter replacement, as described in the main text. Figure S6. Extracellular farnesol production of yeast strain SHEF and SHEF strains expressing LsGAS, AvGAS, and AvGAS-F23W, respectively. Figure S7. Extracellular and intracellular germacrene A accumulation in the yeast strain SHEF after cultivation for 72 h. Figure S8. Location of Phe23 in the modelled structure of AvGAS. The substrate FPP is represented as cyan sticks and Phe23 is shown as purple sticks. Table S1. Growth analyses of yeast cells expressing Phe23 mutants of AvGAS after cultivation for 72 h.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, W., Guo, J., Wang, Z. et al. Improved production of germacrene A, a direct precursor of ß-elemene, in engineered Saccharomyces cerevisiae by expressing a cyanobacterial germacrene A synthase. Microb Cell Fact 20, 7 (2021). https://doi.org/10.1186/s12934-020-01500-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01500-3