
Abstract
Background
Pimobendan has been proven to delay the onset of congestive heart failure (CHF) in dogs with mitral regurgitation (MR); however, molecular underlying mechanisms have not been fully elucidated. This study aimed to investigate (1) the effects of pimobendan on cardiac function, cardiac mitochondrial quality and morphology, and cardiac ultrastructure in a rat model of chronic MR and (2) the direct effect of pimobendan on intracellular reactive oxygen species (ROS) production in cardiac cells. MR was surgically induced in 20 Sprague-Dawley rats, and sham procedures were performed on 10 rats. Eight weeks post-surgery, the MR rats were randomly divided into two groups: the MR group and the MR + pimobendan group. Pimobendan (0.15 mg/kg) was administered twice a day via oral gavage for 4 weeks, whereas the sham and MR groups received equivalent volumes of drinking water. Echocardiography was performed at baseline (8 weeks post-surgery) and at the end of the study (4 weeks after treatment). At the end of the study protocol, all rats were euthanized, and their hearts were immediately collected, weighed, and used for transmission electron microscopy and mitochondrial quality assessments. To evaluate the role of pimobendan on intracellular ROS production, preventive or scavenging properties were tested with H2O2-induced ROS generation in rat cardiac myoblasts (H9c2).
Results
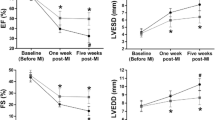
Pimobendan preserved cardiac functions and structure in MR rats. In addition, pimobendan significantly improved mitochondrial quality by attenuating ROS production and depolarization (P < 0.05). The cardiac ultrastructure and mitochondrial morphology were significantly preserved in the MR + pimobendan group. In addition, pimobendan appeared to play as a ROS scavenger, but not as a ROS preventer, in H2O2-induced ROS production in H9c2 cells.
Conclusions
Pimobendan demonstrated cardioprotective effects on cardiac function and ultrastructure by preserving mitochondrial quality and acted as an ROS scavenger in a rat model of MR.
Similar content being viewed by others

Background
Degenerative mitral valve disease (DMVD) is the most common heart disease in dogs responsible for approximately 75% of heart disease cases visiting veterinary practitioners worldwide, especially in North America [1]. In DMVD, the mitral valve does not close properly, causing blood to flow backward into the left atrium from the left ventricle as it contracts. This disease initiates volume overload, leading to several subsequent compensatory mechanisms [2]. Ultimately, these compensatory adjustments weaken the heart and lead to decompensated mitral regurgitation (MR) (i.e., heart failure). In recent prospective studies from randomized clinical trials in the field of veterinary cardiology, pimobendan administration delays the onset of congestive heart failure (CHF) by a median of 15 months in dogs with DMVD stage B2 [3]. Moreover, adding pimobendan to heart failure therapy in dogs with furosemide and angiotensin-converting enzyme inhibitors delayed the onset of refractory signs of heart failure, improved clinical conditions, and increased survival time [4,5,6]. Although pimobendan inhibits phosphodiesterase-III (PDE-III) causing an increase in cyclic adenosine monophosphate (cAMP) and sensitizes the cardiac contractile apparatus (i.e., troponin C) to intracellular calcium, the definite mechanism of pimobendan underlying the delay in the onset of CHF or refractory signs of CHF is still unclear.
Cardiac mitochondrial dysfunction has been demonstrated to play a crucial role in HF and is believed to contribute to the progression of HF through both diminished high-energy phosphate production and amplified production of reactive oxygen species (ROS), resulting in high levels of oxidative stress [7, 8]. The intracellular ROS generated by damaged or malfunctional mitochondria also causes mitochondrial dysfunction, which initiates cardiac remodeling and damages sarcomeric proteins [9]. Furthermore, stretched myocytes produce ROS, leading to cardiomyocyte degeneration, and are associated with contractile dysfunctions in the pathophysiology of HF [20]. Briefly, the incision was made in the fifth intercostal space, and the pericardial sac was cut open. The left auricle was lifted, and the mitral valve was punctured with a 20-G needle through the left ventricular free wall just below the valve location. MR was confirmed by a transthoracic echocardiography machine equipped with a 4–10-MHz phased array probe (M9, Mindray, Shenzhen, China). The MR jet area of 45–60% of the total left atrial area was considered successful and was included in the study (Fig. 1). After that, the thoracic cavity, muscular layer, and skin were closed with an absorbable suture. A similar surgical procedure was performed without puncturing the left ventricle (LV) and mitral leaflet for the sham operation. Enrofloxacin and tramadol were given daily for 7 days. All rats were allowed to develop LA and LV anatomic and molecular remodeling and altered systolic function for 8 weeks before initiating the treatment intervention.
Experimental procedure
Eight weeks after surgery, echocardiography was performed to confirm and document the presence of MR in the MR group, whereas no MR was visible in the sham group. The rats in the MR group were randomly divided into two groups: MR rats (n = 10) receiving drinking water 5 mL/kg, PO, q12h, and MR rats receiving pimobendan (MR + PIMO; n = 10) 0.3 mg/kg, PO, divided twice daily. The dose of pimobendan used in this study was selected based on a previous publication [43] in which chronic administration of pimobendan resulted in an increased EF in catecholamine-induced myocardial injury. Echocardiography was performed to ensure that there were no differences in echocardiographic parameters observed between the two groups at the baseline. The sham group was given drinking water (5 mL/kg, PO, q12h) as a placebo. Echocardiography was conducted again 4 weeks after therapy was initiated (Fig. 9). Four weeks after the initiation of treatment, the rats were sacrificed with an overdose of isoflurane in an anesthetic chamber to harvest their heart. Thoracotomy and vital organ removal (i.e., heart) were used as a physical confirmation of death in the current study. The heart was perfused with ice-cold phosphate-buffered saline (PBS) solution and weighed. A small portion of the LV apex (approximately 300 mg) was collected for mitochondrial isolation, whereas the trabeculae muscle of the left ventricle was obtained for TEM. The remaining part of the heart was placed in 10% formalin for 24 h, and histopathological processes were performed for hematoxylin and eosin staining.

Study design to assess the effects of pimobendan (PIMO) on cardiac function, mitochondrial quality, and cardiac ultrastructure and mitochondrial morphology in rats with mitral regurgitation (MR). Eight weeks after surgical induction of MR, pimobendan (0.15 mg/kg) was given orally to MR + PIMO rats twice daily for 4 weeks, whereas MR rats received drinking water orally (5 mL/kg). Echocardiography images were obtained at baseline (8 weeks after the operation) and at the end of the study (12 weeks after treatment). At the end of the experiments, all hearts were harvested to evaluate their weight per bodyweight ratio. In addition, a heart section was processed for transmission electron microscopy to quantify mitochondrial morphology and sarcomere length. Mitochondrial quality was inferred from three mitochondrial parameters: swelling, membrane potential changes, and reactive oxygen production
Echocardiography
Echocardiography was performed in all rats between 1.5 and 3.0 h post-pimobendan or placebo dosing. Rats were lightly sedated with isoflurane 2–3% mixed with oxygen through the nose cone. Images were acquired with a 4–10-MHz ultrasonic phased array probe connected to a Mindray M9 echocardiography machine, and electrocardiographic (ECG) electrodes were attached to both forelimbs and the left hindlimb and connected to the machine. Color Doppler map** of MR jets was used to semiquantitatively assess the severity of MR. MR jet areas were assessed in the right parasternal long-axis view. LV septal and posterior wall thickness (i.e., interventricular septal at end-diastole, interventricular septal at end-systole, LV posterior wall at end-diastole, and left ventricular posterior wall at end-systole) were measured by M-mode echocardiography at the level of the head of the papillary muscle following previously published guidelines [44]. From this view, fractional shortening, EF, EDV, and ESV were calculated using the Teichholz method [45, 46]. At the base of the heart, a right parasternal short-axis view was obtained to evaluate the left atrium-to-aortic root ratio (LA/Ao). The right parasternal long-axis view with color map** was also obtained to measure the MR jet area (%). The MR jet area (%) was quantified by the area of mosaic color observed during systole inside the left atrium divided by the total area of the left atrium and multiply by 100 which was described previously [47]. All parameters were evaluated on an average of six consecutive cardiac cycles.
Determination of mitochondrial quality: mitochondrial ROS production, membrane potential changes, and swelling
The isolation of cardiac mitochondria was described previously [48]. Briefly, a piece of the LV apex was homogenized with an isolation buffer (300 mM sucrose, 0.2 mM EGTA, 5 mM TES; pH 7.2) in cold conditions using a Dounce homogenizer for the evaluation of mitochondrial function. A differential centrifuge was used to isolate the mitochondria. The mitochondrial pellet was re-suspended with 500 µL of ice-cold respiration buffer (100 Mm, KCl; 50 mM, sucrose; 10 mM, HEPES; 5mM, K2HPO4; pH, 7.2), and the mitochondrial protein concentration was immediately measured using the Bradford protein assay. Bovine serum albumin was used for creating a standard curve and quantifying the concentration equation.
The ROS production by the mitochondria was determined using dichloro-dihydro-fluorescein diacetate (DCFDA) in a fluorescent microplate reader [49]. The mixture of isolated cardiac mitochondria and DCFDA was prepared and added into a black microplate to obtain a final concentration of mitochondria protein of 0.4 mg/mL in a total volume of 200 µL and incubated at 37 °C for 30 min. DCFDA passed into the mitochondria and oxidized in the presence of ROS to form DCF, a fluorescent form. Fluorescence intensity was measured at excitation and emission wavelengths of λ 485 nm and λ 530 nm, respectively.
Cardiac mitochondrial membrane potential changes were determined using 5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) [49]. The diluted JC-1 solution (5 µM) was added into a black microplate, followed by a mitochondrial solution, to obtain 0.4 mg/mL in a total volume of 200 µL and incubated at 37 °C for 30 min in the dark. Fluorescence intensities of green (JC-1 monomer) and red (JC-1 aggregate) were measured at excitation and emission wavelengths of λ 485 and λ 530 nm, and λ 485 and 590 nm, respectively. The red/green intensity ratio was calculated and represented to indicate the quality of mitochondria [50]. A reduction in the red/green fluorescence intensity indicates mitochondrial depolarization.
Mitochondrial swelling was determined by measuring the change in the optical density value at λ540 nm in the respiration buffer (mitochondrial protein concentration of 0.4 mg/mL in a total volume of 200 µL) using a microplate reader [49]. Absorbance was measured immediately and every 5 min after mitochondria were added to the buffer. A rapid loss of the absorbance measured at 30 min indicates mitochondrial swelling.
Visualization of the myocyte ultrastructure by TEM
An LV trabeculae muscle (n = 2/group) was cut and fixed with 3% glutaraldehyde in 0.1 M phosphate buffer and stored at 4 °C for the TEM study. Then, the tissues were postfixed with 2% osmium tetroxide in 0.1 M phosphate buffer, dehydrated with an alcohol series, and embedded in epon (Epon 812; Electron Microscopy Sciences, Fort Washington, PA, USA) and polymerized at 70 °C overnight. Lead citrate and uranyl acetate were used to stain the ultrathin Sects. (65–70 nm). Mitochondrial morphology, sarcomere length, and vacuole size were visualized under a transmission electron microscope (JEM-1400 Plus; JEOL, Tokyo, Japan). Images were captured at low magnification, i.e., 2,500× for sarcomere length assessment and 10,000× for mitochondrial morphology assessment. Sarcomere lengths were measured using NIS-Elements Advance Research (Nikon, USA).
In vitro study of the effect of pimobendan on H2 O2 -induced intracellular ROS production in H9c2 cells.
Cell culture and treatment
The rat ventricular myoblast cell line H9c2 (ATCC number CRL-1446) was purchased from the American Type Culture Collection (ATCC®, Manassas, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (D5030, Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum (FBS-G1-12 A, Biocom Biotech) and 100 U/mL penicillin and 100 µg/mL streptomycin. The cells were incubated in a humidified incubator at 37 °C, under 95% air and 5% CO2. The medium was refreshed every 2 days. Subculture was performed when the cell density reached 80–90% confluence.
The first protocol was designed to obtain the optimum dosage of pimobendan for a subsequent study. The H9c2 cells were seeded at a density of 3 × 103 cells/well in a 96-well plate until reaching 80–90% confluence. Pimobendan (Vetmedin injection, 0.75 mg/mL, MW 334.37) was freshly diluted with fresh media to several concentrations at 0.1, 1, 10, and 100 µM. These ranges of concentration were chosen as they effectively cause augmented glucose-induced insulin release in a dose-dependent manner by sensitizing the intracellular calcium in rats [51]. The cell viability assay was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) cell viability assay, which was evaluated under a microscope after incubating pimobendan for 1, 2, 6, 12, and 24 h. The dosage that did not cause 10% of the individuals to die, or lethal dose 10% (LD10), was chosen for the next protocol. In the second protocol, the effects of pimobendan on the alteration of the mitochondrial membrane potential and intracellular ROS production were determined.
Mitochondrial membrane potential
H9c2 cells were seeded at a density of 3 × 103 cells in 200 µL to a black 96-well plate. Then, 5 µM of JC-1 dye was added and incubated at 37 °C for 30 min in the dark. Fluorescence intensity was determined using a microplate reader. The fluorescence intensity for monomeric green fluorescein was determined by a fluorescence microplate reader with the excitation and emission wavelengths at 485 and 530 nm, respectively, while the aggregate red fluorescein was determined by a fluorescence microplate reader with excitation and emission wavelengths at 485 and 590 nm, respectively.
Intracellular ROS production
H9c2 cells were seeded at a concentration of 5 × 103 cells/mL in 200 µL. Cells were allowed to attach for at least 24 h. After pimobendan treatment, the media was discarded and incubated with 100 µL of complete media containing 5 µM DCFDA. The plate was incubated at 37 °C for 60 min in the dark. To determine ROS production, the fluorescence intensity was determined by a fluorescence microplate ready with excitation and emission wavelengths at 485 and 530 nm, respectively.
Cell viability
H9c2 cells were seeded at a density of 3 × 103 cells in 200 µL until reaching 80% confluence. Then, cells were incubated with a complete medium in the presence and absence of pimobendan and incubated at 37 °C. Cell viability was assessed by the MTT cell viability assay by incubation with 0.5 mg/mL of MTT dye at 37 °C for 2 h. After incubation, the MTT reagent was discarded and dimethylsulfoxide (DMSO) was added for solubilizing the formazan dye. The optical density was determined by a spectrophotometer at λ 490 nm using DMSO as a blank. The relative percentage of cell viability was compared with the control group [52].
Determination of intracellular ROS production by H2O2 challenging
H9c2 cells were seeded at a density of 105 cells in 200 µL until reaching 80% confluence. Pimobendan was freshly prepared by diluting with the fresh media to obtain a concentration of 0, 0.1, 1, 10, and 100 µM. Two protocols were established as an ROS formation preventer and an ROS scavenger.
In the first protocol, to determine the pretreatment effect of pimobendan on intracellular ROS generation, cells were incubated with 25 µM DCFDA in a medium at 37 °C 30 min before incubation with several concentrations of pimobendan. Then, the culture medium was discarded, and cells were treated with PBS or 250 µM H2O2 in PBS for 30 min. The fluorescence signal of DCF was measured at excitation and emission wavelengths of 485 and 530 nm, respectively.
In the second protocol, for determining the ROS-scavenging properties of pimobendan, different concentrations of pimobendan were incubated with PBS (control) or H2O2 to obtain 250 µM H2O2 at 37 °C for 30 min. Before treatment, the cells were incubated with 25 µM DCFDA in a medium for 30 min. Then, the culture medium was replaced by pimobendan–PBS or pimobendan–H2O2 mixtures and incubated at 37 °C for 30 min. The fluorescence signal of DCF was measured at excitation and emission wavelengths of 485 and 530 nm, respectively. The mean fluorescence intensities are expressed as percentages of the untreated control.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Heart-to-bodyweight ratios were calculated for each rat using the data at the end of the study. Statistical analysis was performed using IBM® SPSS® Statistics software (IBM Corp., Armonk, NY, USA). The normal distribution of variables was assessed using the Shapiro–Wilk test. A one-way analysis of variance (ANOVA) test with Tukey’s correction for multiple comparisons was used to evaluate the differences among groups. If the values failed to display normality, ANOVA on ranks was used to evaluate the differences among groups. P-value < 0.05 was considered statistically significant.
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Abbreviations
- ANOVA:
-
analysis of variance
- CO:
-
cardiac output
- cAMP:
-
cyclic adenosine monophosphate
- CHF:
-
congestive heart failure
- DMSO:
-
dimethylsulfoxide
- DMVD:
-
degenerative mitral valve disease
- ECG:
-
electrocardiography
- EDV:
-
end-diastolic volume
- ESV:
-
end-systolic volume
- EF:
-
ejection fraction
- H9c2:
-
cardiac myoblasts
- JC-1:
-
5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide
- LA:
-
left atrium
- LV:
-
left ventricle
- LVIDd:
-
left ventricular internal diameter at end-diastole
- LVIDs:
-
left ventricular internal diameter at end-systole
- MR:
-
mitral regurgitation
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- PBS:
-
phosphate buffered saline
- PDE-III:
-
phosphodiesterase-III
- PIMO:
-
pimobendan
- ROS:
-
reactive oxygen species
- SEM:
-
standard error of the mean
- TEM:
-
transmission electron microscope
References
Keene BW, Atkins CE, Bonagura JD, Fox PR, Haggstrom J, Fuentes VL, et al. ACVIM consensus guidelines for the diagnosis and treatment of myxomatous mitral valve disease in dogs. J Vet Intern Med. 2019;33(3):1127–40.
Gaasch WH, Meyer TE. Left ventricular response to mitral regurgitation: implications for management. Circulation. 2008;118(22):2298–303.
Boswood A, Haggstrom J, Gordon SG, Wess G, Stepien RL, Oyama MA, et al. Effect of Pimobendan in Dogs with Preclinical Myxomatous Mitral Valve Disease and Cardiomegaly: the EPIC Study-A Randomized Clinical Trial. J Vet Intern Med. 2016;30(6):1765–79.
Fuentes VL, Corcoran B, French A, Schober KE, Kleemann R, Justus C. A double-blind, randomized, placebo-controlled study of pimobendan in dogs with dilated cardiomyopathy. J Vet Intern Med. 2002;16(3):255–61.
O’Grady MR, Minors SL, O’Sullivan ML, Horne R. Effect of pimobendan on case fatality rate in Doberman Pinschers with congestive heart failure caused by dilated cardiomyopathy. J Vet Intern Med. 2008;22(4):897–904.
Wess G, Kresken JG, Wendt R, Gaugele J, Killich M, Keller L, et al. Efficacy of adding ramipril (VAsotop) to the combination of furosemide (lasix) and pimobendan (VEtmedin) in dogs with mitral valve degeneration: the VALVE trial. J Vet Intern Med. 2020;34(6):2232–41.
Knowlton AA, Chen L, Malik ZA. Heart failure and mitochondrial dysfunction: the role of mitochondrial fission/fusion abnormalities and new therapeutic strategies. J Cardiovasc Pharmacol. 2014;63(3):196–206.
Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–15.
Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. 2013;112(2):393–405.
Pimentel DR, Amin JK, **ao L, Miller T, Viereck J, Oliver-Krasinski J, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89(5):453–60.
Bayeva M, Ardehali H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr Hypertens Rep. 2010;12(6):426–32.
Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH, et al. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96(5):2247–59.
Cortassa S, Aon MA, Winslow RL, O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J. 2004;87(3):2060–73.
Chattipakorn SC, Thummasorn S, Sanit J, Chattipakorn N. Phosphodiesterase-3 inhibitor (cilostazol) attenuates oxidative stress-induced mitochondrial dysfunction in the heart. J Geriatr Cardiol. 2014;11(2):151–7.
Schenkl C, Heyne E, Doenst T, Schulze PC, Nguyen TD. Targeting mitochondrial metabolism to save the failing heart. Life (Basel). 2023;13(4).
Abbott-Johnson K, Pierce KV, Roof S, Del Rio CL, Hamlin R. Acute Effects of Pimobendan on Cardiac function in Dogs with Tachycardia Induced Dilated Cardiomyopathy: a randomized, Placebo-Controlled, crossover study. Front Vet Sci. 2021;8:646437.
Ishiki R, Ishihara T, Izawa H, Nagata K, Hirai M, Yokota M. Acute effects of a single low oral dose of pimobendan on left ventricular systolic and diastolic function in patients with congestive heart failure. J Cardiovasc Pharmacol. 2000;35(6):897–905.
Pichayapaiboon P, Tantisuwat L, Boonpala P, Saengklub N, Boonyarattanasoonthorn T, Khemawoot P, et al. Pharmacodynamics and pharmacokinetics of Injectable Pimobendan and its metabolite, O-Desmethyl-Pimobendan, in healthy dogs. Front Vet Sci. 2021;8:656902.
Kim KH, Kim YJ, Lee SP, Kim HK, Seo JW, Sohn DW, et al. Survival, exercise capacity, and left ventricular remodeling in a rat model of chronic mitral regurgitation: serial echocardiography and pressure-volume analysis. Korean Circ J. 2011;41(10):603–11.
Jungtanasomboon P, Nussaro S, Winwan H, Suebthawinkul P, Boonpala P, Dong VNK, et al. Vericiguat preserved cardiac function and mitochondrial quality in a rat model of mitral regurgitation. Life Sci. 2023;328:121929.
Tantisuwat L, Saengklub N, Boonpala P, Kumphune S, Panyasing Y, Kalandakanond-Thongsong S, et al. Sacubitril/valsartan mitigates cardiac remodeling, systolic dysfunction, and preserves mitochondrial quality in a rat model of mitral regurgitation. Sci Rep. 2023;13(1):11472.
Ozawa SM, Guzman DS, Hawkins MG, Diao SM, Masri AE, Gunther-Harrington CT, et al. Pharmacokinetics of pimobendan following oral administration to New Zealand White rabbits (Oryctolagus cuniculus). Am J Vet Res. 2022;83(4):356–63.
Yata M, McLachlan AJ, Foster DJ, Hanzlicek AS, Beijerink NJ. Single-dose pharmacokinetics and cardiovascular effects of oral pimobendan in healthy cats. J Vet Cardiol. 2016;18(4):310–25.
Endoh M. Mechanism of action of Ca2 + sensitizers–update 2001. Cardiovasc Drugs Ther. 2001;15(5):397–403.
Saengklub N, Boonyarattanasoonthorn T, Kijtawornrat A, Chantasart D. Preliminary bioequivalence of an oral Pimobendan Solution Formulation with Reference Solution Formulation in Beagle Dogs. Vet Sci. 2022;9(3).
El Sabbagh A, Reddy YNV, Nishimura RA. Mitral valve regurgitation in the contemporary era: insights into diagnosis, management, and future directions. JACC Cardiovasc Imaging. 2018;11(4):628–43.
Reis Filho JR, Cardoso JN, Cardoso CM, Pereira-Barretto AC. Reverse cardiac remodeling: a marker of Better Prognosis in Heart failure. Arq Bras Cardiol. 2015;104(6):502–6.
Ellis-Reis RE, Visser LC, Hsue W, Sharpe AN, Kaplan JL. Echocardiographic evaluation of regurgitant fraction in dogs with subclinical myxomatous mitral valve disease: Method comparison, effects of pimobendan, and reproducibility. J Vet Cardiol. 2023;45:27–40.
Gladden JD, Zelickson BR, Wei CC, Ulasova E, Zheng J, Ahmed MI, et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic Biol Med. 2011;51(11):1975–84.
Guichard JL, Rogowski M, Agnetti G, Fu L, Powell P, Wei CC, et al. Desmin loss and mitochondrial damage precede left ventricular systolic failure in volume overload heart failure. Am J Physiol Heart Circ Physiol. 2017;313(1):H32–H45.
Yancey DM, Guichard JL, Ahmed MI, Zhou L, Murphy MP, Johnson MS, et al. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. Am J Physiol Heart Circ Physiol. 2015;308(6):H651–63.
Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335–44.
Rosado JA, Gonzalez A, Salido GM, Pariente JA. Effects of reactive oxygen species on actin filament polymerisation and amylase secretion in mouse pancreatic acinar cells. Cell Signal. 2002;14(6):547–56.
Ahmed MI, Gladden JD, Litovsky SH, Lloyd SG, Gupta H, Inusah S, et al. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction > 60%. J Am Coll Cardiol. 2010;55(7):671–9.
Chen YW, Pat B, Gladden JD, Zheng J, Powell P, Wei CC, et al. Dynamic molecular and histopathological changes in the extracellular matrix and inflammation in the transition to heart failure in isolated volume overload. Am J Physiol Heart Circ Physiol. 2011;300(6):H2251–60.
Cone J, Wang S, Tandon N, Fong M, Sun B, Sakurai K, et al. Comparison of the effects of cilostazol and milrinone on intracellular cAMP levels and cellular function in platelets and cardiac cells. J Cardiovasc Pharmacol. 1999;34(4):497–504.
Fukasawa M, Nishida H, Sato T, Miyazaki M, Nakaya H. 6-[4-(1-Cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2-(1H)quinolinone (cilostazol), a phosphodiesterase type 3 inhibitor, reduces infarct size via activation of mitochondrial Ca2+-activated K+ channels in rabbit hearts. J Pharmacol Exp Ther. 2008;326(1):100–4.
Corporan D, Segura A, Padala M. Ultrastructural adaptation of the cardiomyocyte to chronic mitral regurgitation. Front Cardiovasc Med. 2021;8:714774.
Li S, Nguyen NUN, **ao F, Menendez-Montes I, Nakada Y, Tan WLW, et al. Mechanism of eccentric cardiomyocyte hypertrophy secondary to severe mitral regurgitation. Circulation. 2020;141(22):1787–99.
Gladden JD, Ahmed MI, Litovsky SH, Schiros CG, Lloyd SG, Gupta H, et al. Oxidative stress and myocardial remodeling in chronic mitral regurgitation. Am J Med Sci. 2011;342(2):114–9.
Prasad K, Gupta JB, Kalra J, Lee P, Mantha SV, Bharadwaj B. Oxidative stress as a mechanism of cardiac failure in chronic volume overload in canine model. J Mol Cell Cardiol. 1996;28(2):375–85.
National Research Council. Guide for the care and use of laboratory animals. 8th ed. Washington D.C.: National Academies Press; 2011.
Nakata TM, Suzuki K, Uemura A, Shimada K, Tanaka R. Contrasting Effects of Inhibition of phosphodiesterase 3 and 5 on cardiac function and interstitial fibrosis in rats with Isoproterenol-Induced Cardiac Dysfunction. J Cardiovasc Pharmacol. 2019;73(3):195–205.
Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American society of Echocardiography Committee on Standards, Subcommittee on quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2(5):358–67.
Reffelmann T, Kloner RA. Transthoracic echocardiography in rats. Evalution of commonly used indices of left ventricular dimensions, contractile performance, and hypertrophy in a genetic model of hypertrophic heart failure (SHHF-Mcc-facp-Rats) in comparison with Wistar rats during aging. Basic Res Cardiol. 2003;98(5):275–84.
Darbandi Azar A, Tavakoli F, Moladoust H, Zare A, Sadeghpour A. Echocardiographic evaluation of cardiac function in ischemic rats: value of m-mode echocardiography. Res Cardiovasc Med. 2014;3(4):e22941.
Kijtawornrat A, Komolvanich S, Saengklub N, Pirintr P, Boonpala P, Buranakarl C. Long-term effect of sildenafil on echocardiographic parameters in dogs with asymptomatic myxomatous mitral valve degeneration. J Vet Med Sci. 2017;79(4):788–94.
Kumphune S, Surinkaew S, Chattipakorn SC, Chattipakorn N. Inhibition of p38 MAPK activation protects cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol. 2015;53(12):1831–41.
Thummasorn S, Kumfu S, Chattipakorn S, Chattipakorn N. Granulocyte-colony stimulating factor attenuates mitochondrial dysfunction induced by oxidative stress in cardiac mitochondria. Mitochondrion. 2011;11(3):457–66.
Sivandzade F, Bhalerao A, Cucullo L. Analysis of the mitochondrial membrane potential using the Cationic JC-1 dye as a sensitive fluorescent probe. Bio Protoc. 2019;9(1).
Fujimoto S, Ishida H, Kato S, Okamoto Y, Tsuji K, Mizuno N, et al. The novel insulinotropic mechanism of pimobendan: direct enhancement of the exocytotic process of insulin secretory granules by increased Ca2+ sensitivity in beta-cells. Endocrinology. 1998;139(3):1133–40.
Prompunt E, Nernpermpisooth N, Sanit J, Kumphune S. Overexpression and pre-treatment of recombinant human secretory leukocyte protease inhibitor (rhSLPI) reduces an in vitro ischemia/reperfusion injury in rat cardiac myoblast (H9c2) cell. Biomol Concepts. 2018;9(1):17–32.
Acknowledgements
The authors would like to thank Chulalongkorn University Laboratory Animal Center (CULAC) for an in-kind contribution.
Funding
This study was supported by Chulalongkorn University Graduate School for the 100th Anniversary Chulalongkorn University Fund for Doctoral Scholarship and the 90th Anniversary of Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund) to P.B. and A.K. (Funding number GCUGR1125641042D). This study was also partly supported by the National budget to CULAC (Funding number 390252002).
Author information
Authors and Affiliations
Contributions
P.B., N.S., and A.K. conceived the project, designed, and performed in vivo animal experiments. P.B., A.K., and S.K. analyzed the results. P.B., N.S., A.K., and S.K. performed mitochondrial quality analysis. S.S., W.J., and P.B. performed transmission electron microscope analysis. P.B. and Y.P. performed histopathology assessment. P.B. and A.K. drafted the manuscript; N.S., S.K., and A.K. supervised and provided critical review of the manuscript. All authors reviewed and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee of Chulalongkorn University Laboratory Animal Center (Animal Use Protocol No. 1873022). All experimental procedures in animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals [42] and Animal Welfare Act 2015. All reported methods are in accordance with the ARRIVE 2.0 guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Boonpala, P., Saengklub, N., Srikam, S. et al. Pimobendan prevents cardiac dysfunction, mitigates cardiac mitochondrial dysfunction, and preserves myocyte ultrastructure in a rat model of mitral regurgitation. BMC Vet Res 19, 130 (2023). https://doi.org/10.1186/s12917-023-03693-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-023-03693-2