
Abstract
Background
Genetic disorders significantly affect patients in neonatal intensive care units, where establishing a diagnosis can be challenging through routine tests and supplementary examinations. Whole-exome sequencing offers a molecular-based approach for diagnosing genetic disorders. This study aimed to assess the importance of whole-exome sequencing for neonates in intensive care through a retrospective observational study within a Chinese cohort.
Methods
We gathered data from neonatal patients at Tian** Children’s Hospital between January 2018 and April 2021. These patients presented with acute illnesses and were suspected of having genetic disorders, which were investigated using whole-exome sequencing. Our retrospective analysis covered clinical data, genetic findings, and the correlation between phenotypes and genetic variations.
Results
The study included 121 neonates. Disorders affected multiple organs or systems, predominantly the metabolic, neurological, and endocrine systems. The detection rate for whole-exome sequencing was 52.9% (64 out of 121 patients), identifying 84 pathogenic or likely pathogenic genetic variants in 64 neonates. These included 13 copy number variations and 71 single-nucleotide variants. The most frequent inheritance pattern was autosomal recessive (57.8%, 37 out of 64), followed by autosomal dominant (29.7%, 19 out of 64). In total, 40 diseases were identified through whole-exome sequencing.
Conclusion
This study underscores the value and clinical utility of whole-exome sequencing as a primary diagnostic tool for neonates in intensive care units with suspected genetic disorders. Whole-exome sequencing not only aids in diagnosis but also offers significant benefits to patients and their families by providing clarity in uncertain diagnostic situations.
Similar content being viewed by others

Background
Genetic disorders (GDs) are a significant concern in the neonatal intensive care unit (NICU), contributing to approximately 20% of infant deaths [1, 2]. The incidence of GDs diagnoses has risen in recent years, largely due to advancements in genomic sequencing [1, 3]. GDs are diverse and have serious clinical implications, affecting patient diagnoses and their quality of life. Early diagnosis is crucial, benefiting from clinical management and treatment. However, clinical signs are often subtle in the early stages, particularly in newborns in the NICU, where symptoms may not be fully apparent. Sometimes, the phenotype of GDs can be obscured by other clinical symptoms, complicating diagnosis despite numerous routine and specialized tests, including invasive procedures and repeated blood sampling. These processes can cause significant distress to patients, financial strain on families, and yet may not elucidate the underlying pathogenesis [4, 5]. The rapid progression of GDs can lead to death or disability if diagnosis and treatment are delayed or missed.
The evolution of molecular diagnostic techniques has significantly enhanced the role of genetic testing in GDs diagnosis [2, 6,7,8]. Recent studies highlight the benefits of genetic testing in clinical settings. For example, a study from China on the largest cohort of neonates with congenital heart defects demonstrated that next-generation sequencing facilitated precise genetic diagnoses, enabling earlier intervention by specialists [9]. A study at Bei**g Children’s Hospital [10] revealed that exome sequencing as an initial test for pediatric respiratory diseases had a diagnostic yield of 34.6%, proving its efficacy in rapidly diagnosing and guiding treatment. Additionally, a prospective study [11] showed that using whole-exome sequencing (WES) as a primary test in infants suspected of monogenic disorders could streamline the diagnostic process, offering a higher diagnostic yield than standard approaches. Recent research has also discussed the application of WES in NICU settings and among neonate populations in China from various perspectives [2, 9, 12, 13], including the study of molecular defects in neonates conceived through assisted reproductive technology. Despite these advancements, the application of WES in diagnosing neonatal genetic diseases warrants further exploration. This paper presents a retrospective observational study on a Chinese cohort of neonates, aiming to discuss the significance of WES for patients in the NICU.
Methods
Recruitment and data collection
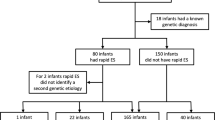
From January 2018 to April 2021, we analyzed 132 neonates hospitalized in the NICU at Tian** Children’s Hospital in China, who presented with acute illness and were suspected of having GDs identified through WES. After excluding 11 cases due to clear etiology, incomplete information, or duplicate collections, 121 cases were included in our study, as illustrated in Fig. 1. The Tian** Children’s Hospital Ethics Committee approved this study, and informed consent was obtained from the guardians or parents. We gathered demographic, prenatal, and intranatal information, along with clinical manifestations, physical examination results, accessory examination outcomes, and family histories of the enrolled patients.

Flow chart of enrolled cases
WES and bioinformatic analysis
Genomic DNA was extracted from the peripheral venous blood of the patients and their parents. WES was conducted by Kingmed Company (Guangzhou, China) and MyGenostics Inc. (Bei**g, China), achieving read lengths of 150 bp and an average coverage depth of 100-200X for over 95% of targeted regions. These regions included the coding areas of more than 20,000 genes and the exon–intron boundaries. Exome capture was performed using the xGen Exome Research Panel v2 (Integrated DNA Technologies, US), and sequencing was carried out on the NovaSeq 6000 system (Illumina, US). The raw data were aligned to the human reference genome hg19 using the Burrows-Wheeler Aligner software. Variant annotations were conducted using the ANNOVAR software, updated bi-monthly, with integration from databases [14] such as RefSeq Gene, dbSNP150, ClinVar, HGMD, and allele frequencies from 1000G, ESP6500, and the ExAC database. Copy Number Variants (CNVs) calling was executed using ExomeDepth software [15] with default settings and batch consistency in sequencing and bioinformatics procedures. The common cause of neonatal hypotonia, homozygous deletion of SMN1, was analyzed using established protocols [16, 17]. Verification of exome results, when necessary, was done through Sanger sequencing and/or Multiplex Ligation-dependent Probe Amplification (MLPA) using the BigDye™ Terminator v3.1 (Applied Biosystems™, US.) on the ABI3530Dx platform (Applied Biosystems™, US), and standard reagents from MRC-Holland (Netherlands) for MLPA.
Pathogenicity assessment
The analysis of genetic reports aimed to explore the connection between phenotype and genetic variation. The pathogenicity of variants was classified according to the criteria set by the American College of Medical Genetics and Genomics (ACMG) [18], which includes five levels: pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign.
Results
Clinical information of neonates in the NICU
This study enrolled 121 neonates, comprising 66 male and 55 female infants, with ages at enrollment ranging from 1 h to 28 days. The majority of the infants were full-term, accounting for 86.8% (105/121), while preterm and extremely preterm infants represented 12.4% (15/121) and 0.8% (1/121), respectively, as detailed in Table 1. The study focused on multiple organ or system involvements, primarily in the metabolic, neurologic, and endocrine systems, with incidence rates of 24% (29/121), 15.7% (19/121), and 12.4% (15/121), respectively, also shown in Table 1. In the metabolic system, the clinical manifestation included poor feeding, vomiting, disturbance of consciousness, metabolic acidosis, hyperammonemia, hyperkalemia, hyperhomocysteinemia, hypotonia, abnormal electroencephalogram, anemia, cardiomyopathy, metabolic alkalosis, hyperphenylalaninemia, lethargy, seizures, coma, poor growth, and intrahepatic cholestasis. The neurologic system's manifestations included conditions such as convulsion, weakness of limbs, hypotonia, loss of tendon reflexes, muscle weakness, inability to suck, axial hypotonia, hearing loss, peculiar hair, weak cry, recurrent bronchopneumonia, swallowing difficulties, respiratory distress, while the endocrine system showed signs such as electrolyte disorders, dark areola, vomiting, poor feeding, and malnutrition were observed in the endocrine system. Additional details on organ involvement are provided in Supplemental Table 1.
Variants information of neonates in the NICU
The overall detection rate of pathogenic or likely pathogenic genomic variants via WES was 52.9% (64/121). We identified 84 genomic variants in 64 neonates, comprising 13 copy number variations (CNVs) and 71 single-nucleotide variants (SNVs), detailed in Table 2. The CNVs mostly involved deletions and duplications, including cases of spinal muscular atrophy and global developmental delay or multiple malformations due to various CNVs. SNVs were primarily missense, nonsense, frameshift, and splicing variations. De novo variations numbered 16 in this cohort, as shown in Table 2. Parental samples were collected simultaneously with the offspring samples for verification by Sanger sequencing upon detection of variant genes. The inheritance patterns are detailed in Fig. 2, with autosomal recessive being the most common (57.8%, 37/64), followed by autosomal dominant (29.7%, 19/64). There was one case of X-linked dominant inheritance, two of X-linked recessive, and five of unknown inheritance patterns. In 4 cases of CNVs, testing at the corresponding loci was recommended for both parents, though it was not performed due to a lack of parental permission. In one case, patient 34 exhibited only one heterozygous pathogenic variant in the FGA gene, suggesting the possibility of undetected mutations by WES.

Proportion of inheritance patterns. Abbreviations: AD, autosomal recessive; AR, autosomal dominant; XLD, X-linked dominant; XLR, X-linked recessive
Genetic disorders diagnosed by WES
WES identified 40 diseases within the study cohort. The most prevalent disorders were methylmalonic acidemia (MMA) (12.5%, 8/64), epilepsy (10.9%, 7/64), spinal muscular atrophy (6.25%, 4/64), and congenital adrenal hyperplasia (6.25%, 4/64), as shown in Fig. 3. Notably, the audiologic system showed a 100% positive rate (3/3), followed by the neurologic system at 68.4% (13/19) in this cohort.

Distribution of diagnosed diseases. Abbreviations: CAH, congenital adrenal hyperplasia; CS, Cohen syndrome; Ep, epilepsy; MMA, methylmalonic acidemia; PA, propionic acidemia; PKU, phenylketonuria; SMA, spinal muscularatrophy; Note: the details of “Other” category shown in Table 2 with superscripted “#”
Discussion
WES has become a primary clinical diagnostic tool for children suffering from developmental delays, intellectual disabilities, respiratory disease [10, 19], and more. Recent discussions have highlighted its use in NICU settings and among neonatal populations in China, viewing it from various perspectives [2, 9, 12, 13]. Despite these discussions, the potential of WES in diagnosing neonatal GDs remains underexplored. In our study, WES was conducted on 121 infants in the NICU at Tian** Children's Hospital of China, yielding a diagnostic rate of 52.9% (64/121). This rate surpasses the 37.9% diagnostic yield of a similar study in the USA [20] and significantly exceeds the 12.3% yield reported in Lin Yang's studies in China [13]. For the discrepancy between this study and two other studies, we analyzed potential reasons based on differences in patient selection and cohort size. Our cohort, selected with stricter inclusion criteria, demonstrated a higher diagnostic yield. We focused on critically ill patients in the NICU, with strong indications for GDs assessed by experienced experts, such as evidence of metabolic disorders identified by mass spectrometry. Additionally, this was a single-center study conducted at Tian** Children’s Hospital, which also accepted critically ill patients from surrounding districts. The cohort size was relatively small, with fewer patients than in the study by Lin Yang et al. [13], which included a cohort of 2,303 neonates in China. In our cohort, a significant proportion of genetically diagnosed patients had metabolic disorders, such as methylmalonic acidemia (MMA), hyperphenylalaninemia, and congenital adrenal hyperplasia. Despite most patients undergoing newborn screening techniques, some reports were negative or showed suspected positive results. In these cases, WES was performed to confirm diagnoses. Among the positive cases, epilepsy and MMA were the most frequently identified diseases, accounting for 10.9% (7/64) and 12.5% (8/64) of cases, respectively.
MMA arising from either a deficiency in methylmalonyl-CoA or abnormal cobalamin metabolism, is a rare, inherited metabolic disorder, primarily passed down through autosomal recessive inheritance. It stands as the most common form of organic acidemia [21]. The disease's genetic underpinnings include mutations in several genes, such as MMACHC, MMADHC, and MMUT, with the prevalence of these mutations varying across different countries and populations [22,23,24]. In China, for instance, the most common mutations in children with MMA are c.609G > A, c.658_660delAAG, and c.80A > G in the MMACHC gene, occurring at frequencies of 34.09%, 13.64%, and 13.64%, respectively [22]. Our study of 8 MMA cases revealed 12 mutations across two genes (MMACHC and MMUT), which included a novel mutation in MMUT c.2131G > T/p.(Glu711*) and eleven inherited mutations. We identified nine gene variations in the MMACHC gene and three mutations in the MMUT gene. The frequency of c.658_660delAAG and c.80A > G mutations was 16.7% (2/12) and 25.0% (3/12), respectively, aligning with findings from previous studies in China [22, 23]. Among eight cases, one was homozygous, while the others were compound heterozygous. All eight MMA patients exhibited an autosomal recessive inheritance pattern. The manifestations of MMA can be nonspecific and vary among patients, especially in newborns and young infants. Previous research has shown that the clinical course of MMA can progress rapidly in neonates, sometimes resulting in death [25, 26] if not treated promptly. Fortunately, MMA is a treatable genetic disorder for most patients [27]. Our team [28] reported a case of a neonate with MMA metabolic decompensation (severe metabolic acidosis and hyperammonemia (> 1,000 µg/dl) who was successfully treated with automatic peripheral arteriovenous exchange transfusion and L-carnitine. The diagnosis was confirmed by WES, leading to a decrease in serum ammonia levels and an improvement in the child’s clinical status. Therefore, WES is crucial for diagnosing this disease, enabling timely treatment and improving the prognosis for NICU patients.
Neonatal seizures are a common manifestation of neurological dysfunction, with an incidence of about 1–5 per 1,000 births [29, 30]. Despite a decrease in mortality from 40 to 20%, the prognosis for neurodevelopmental outcomes, such as cerebral palsy, intellectual disability, and secondary epilepsy, has not significantly improved [30]. Therefore, identifying the cause of neonatal seizures and initiating timely medical treatment is crucial for managing these conditions. The causes of neonatal seizures are diverse, including acute symptomatic seizures, electrolyte imbalances, and cerebral deformity, and so on [30, 31]. Recent advancements in molecular diagnostic technologies, such as WES, have increased the detection rate of genetic disorders causing neonatal seizures [32]. In this study, we identified seven neonatal patients with seizures, uncovering seven mutations in three pathogenic genes. These included five missense mutations, one deletion mutation, and one splicing mutation, comprising one inherited and six de novo variations. For instance, in patient 9, the SCN2A c.781G > A /p.(Val261Met) mutation was identified, leading to a diagnosis of benign familial neonatal convulsions, a form of epilepsy with a favorable prognosis. Thus, WES is valuable for pinpointing genetic causes and guiding precise treatments in NICUs.
In another case, patient 28, a female term infant, was admitted to the NICU of Tian** Children’s Hospital at 25 days old due to jaundice and elevated liver enzymes. The patient, born to a 32-year-old mother, had a normal birth history. Apart from jaundice, the physical examination was unremarkable. Lab tests revealed elevated creatine kinase and glutamic-pyruvic transaminase levels, and an ultrasound cardiogram showed ventricular hypertrophy, suggesting neonatal jaundice, liver dysfunction, and potential hypertrophic cardiomyopathy. Given the unclear etiology, she opted for symptomatic treatment and underwent WES detection. Genetic testing revealed two heterozygous mutations in the GAA gene on chromosome 17q25: c.859-2A > T (p.?) and c.1861 T > G (p.Trp621Gly), confirming the diagnosis of glycogen storage disease type II, also known as Pompe disease. This diagnosis was significantly different from the initial assumption. Pompe disease, a rare autosomal recessive disorder caused by mutations in the GAA gene, leads to a chronic and progressive pathology, predominantly featuring limb-girdle muscle weakness and respiratory failure [33]. Early diagnosis is crucial to mitigate or prevent the irreversible organ damage that progresses with Pompe disease [34]. However, our patient presented without the typical clinical phenotype at admission, posing a diagnostic challenge for clinicians. Thus, WES served as a critical diagnostic tool for patients with unexplained symptoms, ranging from isolated hyper-CKemia to varying degrees of muscular impairment. This case underscores the importance of WES in diagnosing patients with suspected genetic disorders in the NICU, particularly when clinical phenotypes vary widely.
WES analysis presented a negative diagnostic yield of 47.1% (57/121) within this cohort, potentially constrained by the limitations inherent to WES. While WES can detect a wide array of variants, it has a restricted capability in identifying non-coding region variants, abnormal genomic structures, and genomic methylation [35, 36]. However, WES offers significant advantages. Firstly, it covers a broad range of detection and has become more affordable, making it accessible to most parents. Secondly, WES is invaluable for precise diagnosis and treatment strategies, potentially increasing the number of diagnosed infants in NICUs with GDs, thereby reducing infant mortality and morbidity through early neonatal diagnosis. Lastly, WES plays a crucial role in genetic counseling for parents of infants with GDs, enabling informed reproductive decisions. As a vital supplement to standard diagnostics, WES is indispensable for diagnosing GDs in NICU patients.
Conclusions
In conclusion, our findings underscore the critical role of WES in uncovering the etiology, offering targeted therapy, and enhancing the prognosis for patients with suspected GDs in NICUs, especially when diagnoses are complicated by diverse clinical phenotypes. Reflecting on this study and the evidence gathered from this cohort, we advocate for WES as the primary testing approach for suspected GDs cases in NICUs. This recommendation aligns with the evolving trend towards precision medicine, highlighting WES's clinical utility and its importance to patients and their families in cases lacking a clear diagnosis.
Availability of data and materials
All data generated or analyzed in this study are included in this published article.
Abbreviations
- GDs:
-
Genetic disorders
- NICU:
-
Neonatal intensive care unit
- WES:
-
Whole-exome sequencing
- CNVs:
-
Copy number variations
- MLPA:
-
Multiplex ligation-dependent probe amplification
- SNVs:
-
Single-nucleotide variants
- MMA:
-
Methylmalonic acidemia
References
Kingsmore SF, Henderson A, Owen MJ, Clark MM, Hansen C, Dimmock D, et al. Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. NPJ Genom Med. 2020;5:49.
Yang L, Liu X, Li Z, Zhang P, Wu B, Wang H, et al. Genetic aetiology of early infant deaths in a neonatal intensive care unit. J Med Genet. 2020;57(3):169–77.
Wang H, Lu Y, Dong X, Lu G, Cheng G, Qian Y, et al. Optimized trio genome sequencing (OTGS) as a first-tier genetic test in critically ill infants: practice in China. Hum Genet. 2020;139:473–82.
Elliott AM, du Souich C, Lehman A, Guella I, Evans DM, Candido T, et al. RAPIDOMICS: rapid genome-wide sequencing in a neonatal intensive care unit-successes and challenges. Eur J Pediatr. 2019;178:1207–18.
Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10.
Lu JT, Ferber M, Hagenkord J, Levin E, South S, Kang HP, et al. Evaluation for Genetic Disorders in the Absence of a Clinical Indication for Testing: Elective Genomic Testing. J Mol Diagn. 2019;21:3–12.
Sanford EF, Clark MM, Farnaes L, Williams MR, Perry JC, Ingulli EG, et al. Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU. Pediatr Crit Care Med. 2019;20:1007–20.
Ouyang X, Zhang Y, Zhang L, Luo J, Zhang T, Hu H, et al. Clinical Utility of Rapid Exome Sequencing Combined With Mitochondrial DNA Sequencing in Critically Ill Pediatric Patients With Suspected Genetic Disorders. Front Genet. 2021;12: 725259.
Wang H, **ao F, Qian Y, Wu B, Dong X, Lu Y, et al. Genetic architecture in neonatal intensive care unit patients with congenital heart defects: a retrospective study from the China Neonatal Genomes Project. J Med Genet. 2023;60:247–53.
Hao C, Guo R, Liu J, Hu X, Guo J, Yao Y, et al. Exome sequencing as the first-tier test for pediatric respiratory diseases: A single-center study. Hum Mutat. 2021;42:891–900.
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–6.
Huang Z, **ao F, **ao H, Lu Y, Yang L, Zhuang D, et al. Comparison of Genetic Profiles of Neonates in Intensive Care Units Conceived With or Without Assisted Reproductive Technology. JAMA Netw Open. 2023;6: e236537.
Yang L, Wei Z, Chen X, Hu L, Peng X, Wang J, et al. Use of medical exome sequencing for identification of underlying genetic defects in NICU: Experience in a cohort of 2303 neonates in China. Clin Genet. 2022;101:101–9.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38: e164.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformormatics. 2012;28:2747–54.
Larson JL, Silver AJ, Chan D, Borroto C, Spurrier B, Silver LM. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet. 2015;16:100.
Zhao S, Wang Y, **n X, Fang Z, Fan L, Peng Z, et al. Next generation sequencing is a highly reliable method to analyze exon 7 deletion of survival motor neuron 1 (SMN1) gene. Sci Rep. 2022;12:223.
Richands S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
**ang J, Ding Y, Yang F, Gao A, Zhang W, Tang H, et al. Genetic Analysis of Children With Unexplained Developmental Delay and/or Intellectual Disability by Whole-Exome Sequencing. Front Genet. 2021;12: 738561.
Powis Z, Farwell Hagman KD, Speare V, Cain T, Blanco K, Mowlavi LS, et al. Exome sequencing in neonates: diagnostic rates, characteristics, and time to diagnosis. Genet Med. 2018;20:1468–71.
Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C. Methylmalonic and propionic acidemia. Am J Med Genet C Semin Med Genet. 2006;142C:104–12.
Wang C, Liu Y, Zhang X, Wang H, Cui Y, Zhi X, et al. Phenotypic and genotypic analysis of children with methylmalonic academia: A single-center study in China and a recent literature review. Clin Chim Acta. 2021;522:14–22.
Hu S, Mei S, Liu N, Kong X. Molecular genetic characterization of cblC defects in 126 pedigrees and prenatal genetic diagnosis of pedigrees with combined methylmalonic aciduria and homocystinuria. BMC Med Genet. 2018;19:154.
Wang C, Liu Y, Cai F, Zhang X, Xu X, Li Y, et al. Rapid screening of MMACHC gene mutations by high-resolution melting curve analysis. Mol Genet Genomic Med. 2020;8: e1221.
Han L, Wu S, Ye J, Qiu W, Zhang H, Gao X, et al. Biochemical, molecular and outcome analysis of eight chinese asymptomatic individuals with methyl malonic acidemia detected through newborn screening. Am J Med Genet A. 2015;167A:2300–5.
Fischer S, Huemer M, Baumgartner M, Deodato F, Ballhausen D, Boneh A, et al. Clinical presentation and outcome in a series of 88 patients with the cblC defect. J Inherit Metab Dis. 2014;37:831–40.
Wang SJ, Zhao YY, Yan CZ. Reversible encephalopathy caused by an inborn error of cobalamin metabolism. Lancet. 2019;393: e29.
Cui X, Li N, Xue H, Zhang F, Shu J, Liu Y. Case report: Is exchange transfusion a possible treatment for metabolic decompensation in neonates with methylmalonic aciduria in the setting of limited resources? Front Pediatr. 2022;10: 926793.
Shellhaas RA. Seizure classification, etiology, and management. Handb Clin Neurol. 2019;162:347–61.
Pisani F, Spagnoli C, Falsaperla R, Nagarajan L, Ramantani G. Seizures in the neonate: A review of etiologies and outcomes. Seizure. 2021;85:48–56.
Soul JS. Acute symptomatic seizures in term neonates: Etiologies and treatments. Semin Fetal Neonatal Med. 2018;23:183–90.
Spoto G, Saia MC, Amore G, Gitto E, Loddo G, Mainieri G, et al. Neonatal Seizures: An Overview of Genetic Causes and Treatment Options. Brain Sci. 2021;11:1295.
Taverna S, Cammarata G, Colomba P, Sciarrino S, Zizzo C, Francofonte D, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020;12:15856–74.
Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatment make a difference. Pediatr Neonatol. 2013;54:219–27.
Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:2029–37.
Dan H, Huang X, **ng Y, Shen Y. Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol Genet Genomic Med. 2020;8: e1131.
Acknowledgements
Not applicable.
Funding
This work was supported in part by grant 21JCYBJC00370 from Tian** Municipal Science and Technology Bureau, grant TJWJ2021ZD007 from the Public Health and Technology Project of Tian**, and grant 21JCZDJC00390 from the Program of Tian** Science and Technology Plan and grant Y2020003 from Key Project of Tian** Children’s Hospital and grant 21JCZDJC01030 from Natural Science Foundation of Tian**. This study was also funded by Tian** Key Medical Discipline (Specialty) Construction Project (grant number TJYXZDXK-040A).
Author information
Contributions
All authors contributed to this manuscript. Y.L., C.C., and J.S. conceived and supervised the study. R.Z. conceived the study, analyzed the data, and wrote the manuscript. X.C., H.M., and J.G. conducted and analyzed the data. Y.Z. and Y.Z. collected and acquired the clinical data. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Tian** Children’s Hospital, and written informed consent was obtained from the guardians or parents.
Consent for publication
No individual consent for publication is required for this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, R., Cui, X., Zhang, Y. et al. Whole-exome sequencing as the first-tier test for patients in neonatal intensive care unit: a Chinese single-center study. BMC Pediatr 24, 351 (2024). https://doi.org/10.1186/s12887-024-04820-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04820-0