
Abstract
Background
Charcot-Marie-Tooth (CMT) disease is a group of inherited peripheral neuropathies, which are subdivided into demyelinating and axonal forms. Biallelic mutations in POLR3B are the well-established cause of hypomyelinating leukodystrophy, which is characterized by hypomyelination, hypodontia, and hypogonadotropic hypogonadism. To date, only one study has reported the demyelinating peripheral neuropathy phenotype caused by heterozygous POLR3B variants.
Case presentation
A 19-year-old male patient was referred to our hospital for progressive muscle weakness of the lower extremities. Physical examination showed muscle atrophy, sensory loss and deformities of the extremities. Nerve conduction studies and electromyography tests revealed sensorimotor demyelinating polyneuropathy with secondary axonal loss. Trio whole-exome sequencing revealed a de novo variant in POLR3B (c.3137G > A).
Conclusions
In this study, we report the case of a Chinese patient with a de novo variant in POLR3B (c.3137G > A), who manifested demyelinating CMT phenotype without additional neurological or extra-neurological involvement. This work is the second report on POLR3B-related CMT.
Similar content being viewed by others

Background
Charcot-Marie-Tooth (CMT) disease is a group of inherited peripheral neuropathies clinically characterized by progressive distal muscle weakness and atrophy, sensory loss, areflexia and skeletal deformities. By virtue of electrophysiological results, it is classified into two main subgroups: demyelinating form (CMT1) and axonal form (CMT2). Biallelic mutations in POLR3B are the well-established cause of hypomyelinating leukodystrophy, typically featured by hypomyelination, hypodontia, and hypogonadotropic hypogonadism [1, 2]. A recent study reported six unrelated patients carrying heterozygous de novo variants in POLR3B manifested ataxia, developmental delay, spasticity and demyelinating neuropathy, which differed from previously reported phenotypes of POLR3B-related hypomyelinating leukodystrophy [3]. Notably, five of the six patients exhibited predominant demyelinating sensory and motor neuropathy, which unveiled for the first time a demyelinating peripheral neuropathy phenotype caused by POLR3B variants. However, this study has not been further validated by other studies. Here, we report a Chinese patient carrying a de novo variant in POLR3B (c.3137G > A) presenting with demyelinating CMT phenotype without other neurological or extra-neurological involvement.
Case presentation
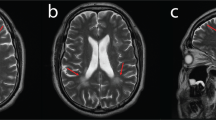
The proband was a 19-year-old man born after an uneventful pregnancy with no family history of neurologic disease. He was born at 39 weeks with a low birth weight of 2350 g. The patient started walking at around 12 months of age with no delay in achieving early developmental milestones. At 5 years old, he developed mild muscle weakness in both legs and fell frequently. Around the age of 7 years, the patient manifested symmetrical muscle atrophy of the distal lower extremities. As the condition progressed, muscle atrophy in the lower extremities worsened and extended to the upper limbs. The physical examination performed at age 8 revealed mild pes cavus and steppage gait. The bilateral knee reflexes were normal, but the ankle reflexes were absent. At 13 years old, symptoms of progressive gait deterioration and dysesthesia in the distal lower limbs were noted. At 19 years old, physical examinations revealed normal cognition, cranial nerves, and cerebellar functions, but obvious muscle atrophy in the distal muscle, pes cavus and claw hands (Fig. 1 A). Motor examination revealed 4/5 muscle strength in proximal limb muscles and wrist extensors, 3/5 in distal muscles of the lower limbs except in ankle dorsiflexors and evertors (0/5). Sensory examination showed decreased pinprick and vibration sensation in the distal extremities. Routine blood count, liver, renal and thyroid functions, blood glucose, and serum electrolyte were normal. Nerve conduction studies (NCS) and needle electromyography (EMG) tests were performed in his 8, 15 and 19 years old, and revealed progressive worsening of sensorimotor demyelinating polyneuropathy with secondary axonal loss (NCS result at the age of 8 was shown in Table 1, no response could be elicited in NCS at the age of 15 and 19). Sural nerve biopsy revealed severe loss of myelinated nerve fibers (Fig. 1B). Brain magnetic resonance imaging (MRI) revealed no specific abnormalities, including corpus callosum, cerebellum, or periventricular white matter (Fig. 1 C).

The clinical manifestations the patient. (A) Neurological examinations showed atrophy of the distal muscles in the extremities and pes cavus. (B) Sural nerve biopsy revealed a significant reduction in myelinated nerve fiber density. Hematoxylin and eosin staining (upper-left penal), Congo red staining (upper-right penal), myelin basic protein staining (lower-left penal), semithin sections (lower-right penal). (C) The brain MRI images of the patient
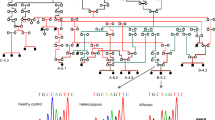
The patient was diagnosed with CMT based on the combination of typical progressive CMT clinical features, sural biopsy result and EMG presentation. The trio whole-exome sequencing (WES) of the proband and his parents was performed on an Illumina NovaSeq6000® in 2019, but no candidate variant of known CMT causative genes was detected at the time. The multiplex ligation-dependent probe amplification (MPLA) analysis was performed and copy number variants in PMP22, MPZ and GJB1 were excluded. Reanalysis of the data in 2021 revealed a candidate variant in POLR3B gene (c.3137G > A, p.R1046H, Fig. 2 A, B), which was reported early this year to be associated with a different set of clinical features including demyelinating neuropathy. The variant was de novo, which was confirmed by parenthood analysis (Fig. 2 A). The variant was absent from the ExAC, dbSNP, 1000G, gnomAD and our WES database which contains 1000 Chinese controls. The Arg1046 residue is highly conserved throughout vertebrate species (Fig. 2 C), suggesting that the mutation may be damaging. Also, it was predicted to be deleterious by SIFT, Polyphen-2, M-CAP, REVEL and ClinPred.

The genetic findings of the patient. (A) The genetic examination revealed that the patient carried a de novo variant (POLR3B: c.3137G > A, p.R1046H), and the parent-child relationship was established by parenthood analysis. (B) Chromatograms of the target variant. (C) Conservation analysis of amino acid sequences on p. R1046H variant sites are highly conserved
Considering hypogonadotropic hypogonadism caused by biallelic variants in POLR3B, serum reproductive hormone levels, including testosterone (TST), follicle stimulating hormone (FSH), luteinizing hormone (LH), estradiol (E2) and progesterone (PRG), were detected and were within normal range. Oral examination revealed no obvious abnormalities.
Discussion and Conclusions
In our study, the first diagnosis of the patient was CMT according to his typical clinical manifestation and clinical examination results. Distal myopathies were ruled out because of severe sensory nerve involvement in the NCS and biopsy. Systemic disorders with neuropathy were ruled out because no significant central nervous system involvement or other generalized symptoms were observed and all simple blood examinations were normal [4, 5]. Early-onset pes cavus with mild sensory symptoms made the acquired demyelinating neuropathy such as CIDP unlikely. Though the clinical diagnosis was explicit, the genetic cause was not known until we reanalyzed the genetic data this year. Thus, POLR3B should be included in genetic testing panel for CMT. In addition, variants of uncertain significance should be routinely reevaluated considering their high reclassification rate and broader phenotypic spectrum of the corresponding genes.
Recessive mutations in POLR3B are recognized to cause hypomyelinating leukodystrophy. Interestingly, Djordjevic et al. have recently reported that heterozygous de novo variants in POLR3B are also pathogenic and cause a broad clinical phenotypic spectrum, including demyelinating neuropathy, seizure and spasticity [3]. In their study, one patient had the same variant (POLR3B c.3137G > A) and similar phenotype as our patient. Different from the other five patients in their study, the patient carrying this variant manifested predominantly demyelinating polyneuropathy without any additional neurological symptoms, such as intellectual disability, developmental delay, language difficulties, seizure and spasticity. The only difference between the two patients was that their patient showed Chiari type I malformation, which was more likely a comorbidity than a developmental consequence of the causative variant in POLR3B. The similar phenotypes of patients carrying the same variant (POLR3B c.3137G > A) may indicate a potential relationship between different genotypes and various phenotypes, which needs to be explored by further extended studies. Notably, there was no overlap among recessive and de novo mutations in POLR3B.
POLR3B encodes the second largest subunit of RNA polymerase III, which is involved in transcription of small non-coding RNA. Using affinity purification coupled with mass spectrometry (AP-MS), Djordjevic et al. revealed POLR3B c.3137G > A distinctly affected normal assembly of RNA polymerase III. Due to technical and material limitations, we did not perform functional studies on this variant, which was a limitation of our study. In addition, clinical symptoms and neurological examination results may evolve as the disease progress, and it is important to continue follow-up study.
Collectively, we report an early-onset demyelinating CMT case carrying a de novo variant in POLR3B, the second global report involving POLR3B-related CMT.
Availability of data and materials
Not applicable.
Abbreviations
- CMT:
-
Charcot-Marie-Tooth
- MRI:
-
Magnetic resonance imaging
- NCS:
-
Nerve conduction studies
- EMG:
-
Electromyography
- WES:
-
Whole-exome sequencing
- MLPA:
-
Multiplex ligation-dependent probe amplification
References
Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83:1898–905.
Richards MR, Plummer L, Chan YM, Lippincott MF, Quinton R, Kumanov P, et al. Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet. 2017;54:19–25.
Djordjevic D, Pinard M, Gauthier MS, Smith-Hicks C, Hoffman TL, Wolf NI, et al. De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy. Am J Hum Genet. 2021;108:186–93.
Azhary H, Farooq MU, Bhanushali M, Majid A, Kassab MY. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81:887–992.
Pareyson D. Differential diagnosis of Charcot-Marie-Tooth disease and related neuropathies. Neurol Sci. 2004;25:72–82.
Acknowledgements
We would like to thank the participant for his support and willingness to participate in this study.
Funding
This study was supported by the Natural Science Foundation of Zhejiang Province to Hao Yu (LQ19H090017). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YYX: data acquisition, analysis and interpretation of the data, and drafting the manuscript; HLC: data acquisition, analysis and interpretation of the data; HLD and HMY: data acquisition and literature search; YY and LCM: data acquisition and interpretation; ZYW: manuscript review; HY: concept, design, definition of intellectual content, and manuscript review. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Ethical Committee of Second Affiliated Hospital, Zhejiang University School of Medicine, and the patient signed written informed consent before obtain the data.
Consent for publication
We obtained signed consent from the patient for the personal or clinical details to be published in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, YY., Cheng, HL., Dong, HL. et al. A de novo variant of POLR3B causes demyelinating Charcot-Marie-Tooth disease in a Chinese patient: a case report. BMC Neurol 21, 402 (2021). https://doi.org/10.1186/s12883-021-02399-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-021-02399-y