
Abstract
The taxonomic classification of Picea meyeri and P. mongolica has long been controversial. To investigate the genetic relatedness, evolutionary history, and population history dynamics of these species, genoty**-by-sequencing (GBS) technology was utilized to acquire whole-genome single nucleotide polymorphism (SNP) markers, which were subsequently used to assess population structure, population dynamics, and adaptive differentiation. Phylogenetic and population structural analyses at the genomic level indicated that although the ancestor of P. mongolica was a hybrid of P. meyeri and P. koraiensis, P. mongolica is an independent Picea species. Additionally, P. mongolica is more closely related to P. meyeri than to P. koraiensis, which is consistent with its geographic distribution. There were up to eight instances of interspecific and intraspecific gene flow between P. meyeri and P. mongolica. The P. meyeri and P. mongolica effective population sizes generally decreased, and Maxent modeling revealed that from the Last Glacial Maximum (LGM) to the present, their habitat areas decreased initially and then increased. However, under future climate scenarios, the habitat areas of both species were projected to decrease, especially under high-emission scenarios, which would place P. mongolica at risk of extinction and in urgent need of protection. Local adaptation has promoted differentiation between P. meyeri and P. mongolica. Genotype‒environment association analysis revealed 96,543 SNPs associated with environmental factors, mainly related to plant adaptations to moisture and temperature. Selective sweeps revealed that the selected genes among P. meyeri, P. mongolica and P. koraiensis are primarily associated in vascular plants with flowering, fruit development, and stress resistance. This research enhances our understanding of Picea species classification and provides a basis for future genetic improvement and species conservation efforts.
Similar content being viewed by others
Introduction
Plants have extensive capacities for adaptation to local environments [1], and positive selection is a key driver of this local adaptation, leaving traces of selective sweeps on nearby genes linked to those that are selected [2]. Ecological specialization through local adaptation can lead to the emergence of new species, and this ecologically adaptive differentiation has been an important mechanism underlying the emergence of many species [3, 4]. In heterogeneous environments, locally specific climate variables can exert selection pressure on plants, driving adaptive differentiation [5]. However, many species in nature are still undergoing differentiation [6, 7]; thus, populations of the same species that are distributed in different regions are often mistakenly identified as different subspecies or species due to minor morphological differences [8]. The genus Picea is an important component of northern and subalpine forests, with approximately 16 species and 9 varieties in China alone [9]. Due to incomplete reproductive isolation between species, frequent gene flow, and a tendency toward reticulate evolution [10], the interspecific relationships and evolutionary history within the genus Picea have become complex, and the classification of some Picea species is controversial. Therefore, understanding gene flow between species and how the environment drives adaptive evolution is crucial for revealing the mechanisms of species differentiation and classification within the genus Picea.
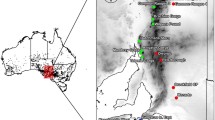
P. meyeri and P. mongolica are unique species of the genus Picea that grow in China. P. meyeri is primarily found in locations such as Guandi Mountain, Wutai Mountain, and Wuling Mountain, while P. mongolica is mainly distributed in the eastern part of the Hunshandake Desert, occupying a relatively narrow range and serving as a component of rare sandy forest grasslands. Due to their similar morphologies and neighborhood distributions, these species are ideal study organisms for understanding Picea differentiation mechanisms. The renowned Chinese taxonomist Zheng Wanjun [11] considered the spruce forest in Bayanaobo, Inner Mongolia, to be a P. koraiensis forest. However, in 1983, Xu [12] argued on the basis of morphological identification and geographical distribution data that this forest was composed of P. meyeri. Wu [13] later compared the Picea species found in this forest with P. meyeri and P. koraiensis and proposed it to be a local variety of P. meyeri, named P. meyeri var. mongolica, which was later supported by Li et al. [ In this study, a total of 223 leaf samples were collected from five species within the genus Picea. The samples were obtained from 130 individuals belonging to 9 natural populations of P. meyeri and 58 individuals belonging to 4 natural populations of P. mongolica, covering most of the distribution areas of the two species. Additionally, the study included 25 individuals from 2 natural populations of P. koraiensis, 5 individuals from 1 natural population of P. likiangensis, and 5 individuals from 1 seed garden population of P. pungens (Fig. 1, Table S1). The leaves of P. meyeri, P. mongolica, and P. koraiensis are all quadrangular-linear. The apexes of the P. meyeri and P. mongolica leaves are acute, while those of the P. koraiensis leaves are obtuse. The leaves of P. mongolica are covered with a white powder that fills the stomatal lines, which are less pronounced in P. meyeri and P. koraiensis. The number of stomatal lines on the leaves decrease in the following order: P. mongolica, P. meyeri, and P. koraiensis. The cones of P. mongolica are larger than those of P. meyeri and P. koraiensis [15] (Fig. 2). The individuals sampled in each natural population were distributed approximately 100 m apart. The collected leaves were immediately dried with silica gel and stored at low temperature. The longitude, latitude, and altitude of each population were recorded using an eTrex handheld GPS device (Garmin, Germany). All these samples were identified by Junhui Wang, who is a professor at the Research Institute of Forestry, Chinese Academy of Forestry. All voucher specimens were deposited at the Research Institute of Forestry, Chinese Academy of Forestry, and detailed sample information is provided in Table S1. Distribution of sampling sites for P. meyeri, P. mongolica and P. koraiensis. Kor1 and Kor2: P. koraiensis from Mengke Mountain and Linjiang, respectively; Mey1, Mey2, Mey3, Mey4, Mey5, Mey6, Mey7, Mey8, and Mey9: P. meyeri from Saihanba, Kelan, Wuzhai, Shenchi, Ningwu, Jiaocheng, Wutai Mountain, **aoWutai Mountain, and Wuling Mountain, respectively; Mon1, Mon2, Mon3, and Mon4: P. mongolica from Baiyinaobao, Dajuzi, Huanggangliang, and Huamugou, respectively Phenotype photographs of P. meyeri, P. mongolica and P. koraiensis. A, B and C are the branches of P. meyeri, P. mongolica and P. koraiensis, respectively; D, E and F are the leaves of P. meyeri, P. mongolica and P. koraiensis, respectively In this study, DNA was extracted from leaves using a modified cetyltrimethylammonium bromide (CTAB) method [22]. The concentration and integrity of the DNA were determined using a NanoDrop 1000 spectrophotometer (Nanodrop, MA, USA) and 1% agarose gel electrophoresis, respectively. Following DNA extraction, sequencing was performed using the double-digest GBS technique [23, 24]. For each sample, after extracting 1.5 µg of DNA, digestion was initially conducted with the EcoRI and NiaIII restriction enzymes, followed by the ligation of adaptors to both ends of the DNA fragments using T4 DNA ligase (NEB) and subsequent amplification. DNA fragments ranging from 400 to 600 bp were then recovered and purified using 1% agarose gel electrophoresis. Finally, the purified products were sequenced on the Illumina HiSeq 4000 sequencing platform (Illumina, CA, USA) using 150-bp paired-end sequencing. For SNP calling, the raw data were filtered using Fastp v0.23.4 [25], in which the 5’-end adaptor sequences, anomalous nucleotide bases, and low-quality ends of the reads were removed (reads with ≥ 10% identified nucleotides and reads with > 50% bases having Phred quality scores ≤ 10). We aligned the filtered clean data for each Picea sample to the P. abies genome using Burrows–Wheeler Aligner (BWA) v0.7.8-r45 [26]. SNP calling was then performed using the Bayesian method in Genome Analysis Toolkit (GATK) v4 [27], followed by preliminary filtering of SNPs based on variant components to generate the SNP dataset. For subsequent phylogenetic and population analyses, the SNP dataset was filtered using VCFtools v0.1.11 [28] (minimum allele frequency ≥ 0.02, maximum missing rate ≤ 0.5) to obtain high-quality SNPs (Dataset I). Using the whole-genome SNP dataset (Dataset I), a maximum likelihood (ML) tree was constructed in IQ-TREE v1.6.10 [29] to explore the phylogenetic relationships among species, setting the number of bootstrap iterations to 1000. Based on the Bayesian information criterion (BIC), the optimal phylogenetic tree model was selected using ModelFinder in IQ-TREE v1.6.10 [29], with the GTR + I model identified as the best model. However, considering that phylogenetic tree reconstruction was based on SNPs, the GTR + I + ASC model was ultimately chosen. The phylogenetic tree results were visualized using iTOL v5 [30]. Admixture v1.3.0 [31] was used to assess population structure. The SNP data (Dataset I) were formatted and pruned for linkage disequilibrium using VCFtools v0.1.11 and PLINK v1.90 (indep-pairwise 50 10 0.1). The number of population clusters (K) ranged from 2 to 10, with 1000 bootstrap replicates employed. The individual genetic composition coefficient (Q) of each group was used to construct a population genetic structure matrix, and cross-validation (CV) error was employed to explore convergence and determine the optimal number of clusters. The population genetic structure matrix was visualized using the Pophelper v2.3.1 package in R v4.3.1. Population genetic structure was analyzed using principal component analysis (PCA) with GCTA v1.93.3 [32] based on SNPs (Dataset I). The results were visualized using the ggplot2 v3.4.3 package in R v4.3.1. Nucleotide diversity (π), Tajima’s D, and population genetic differentiation (FST) were calculated with the PopGenome v2.7.5 package in R v4.3.1. The observed heterozygosity (Ho) and expected heterozygosity (He) were computed using PLINK v1.90 with the default settings. Visualization of the PCA and FST results was conducted using the ggplot2 v3.4.3 package in R v4.3.1. To detect gene flow among P. meyeri, P. mongolica, and P. koraiensis, TreeMix v1.13 [33] and Dsuite v0.5-r44 [34] were used for TreeMix analysis and ABBA–BABA analysis, respectively. To minimize the effects of linkage disequilibrium, SNPs (Dataset I) were filtered (indep-pairwise 50 10 0.1) using PLINK v1.90 [35] to obtain Dataset II for gene flow analysis. In the TreeMix analysis, the migration parameter (m) values were set from 1 to 10, with 5 repetitions for each m value. We utilized the OptM v0.1.6 package in R v4.3.1 to determine the optimal number of migration edges. Subsequently, visualization was conducted using the built-in R script “plotting_funcs.R” in TreeMix v1.13. In the ABBA–BABA analysis, Patterson’s D-statistic was computed using the Dtrios program from Dsuite v0.5-r44. P. pungens and P. likiangensis were used as outgroups (O), P. koraiensis was designated the ancestral group (P1), P. meyeri was designated sister group I (P2), and P. mongolica was designated sister group II (P3). In summary, for the {O, [P1, (P2, P3)]} combination, the ABBA site pattern refers to the sharing of derived alleles between P1 and P2, while the BABA site pattern refers to the sharing of derived alleles between P1 and P3. Under the null hypothesis of incomplete lineage sorting, the numbers of ABBA and BABA site patterns should be roughly equal (D = 0). A significant deviation of D from 0 indicates the occurrence of gene flow events. The significance of the ABBA–BABA result was determined by using a jackknife method to calculate the Z score (Z score > 3). Historical population dynamics were analyzed using Stairway Plot v2.2.1 [36] based on the site frequency spectrum (SFS) of SNPs (Dataset I), where the SFS was computed using the Python v3.9 script easySFS (https://github.com/isaacovercast/easySFS). The SFS state was designated “folded” to calculate the SFS of the second allele. Projection values with as many selected loci as possible were chosen to output SFS information. The SFS information was input into the required blueprint file. The generation time was set at 50 years, and the per-generation neutral mutation rate was set at 2.5 × 10−8 [37, 38]. Moreover, the number of randomly selected loci was set to 67%, and 200 inputs were used to estimate the median Ne and 95% pseudoconfidence interval (CI). The point data for P. meyeri and P. mongolica distribution were primarily sourced from the Chinese Virtual Herbarium (CVH, http://www.cvh.ac.cn/), the National Specimen Information Infrastructure of China (http://www.nsii.org.cn), and published literature [39]. After filtering through ENM Tools v1.4, 77 effective distribution points for P. meyeri and 31 points for P. mongolica were retained (Table S2). The data for nineteen environmental factors were downloaded from the WorldClim database (http://www.worldclim.org), with a resolution of 2.5 arc min. To avoid overfitting the species distribution models (SDMs) due to multicollinearity, Pearson correlation coefficients between the environmental factors were calculated; for variables with |r| > 0.7, the variable with the smaller contribution (estimated through a jackknife test) was removed. The Kuenm v1.1.3 package in R v3.6.3 was used to select the best feature types and regularization multipliers. The Maxent v3.4.4 model was applied to run the SDMs for the two Picea species during four periods (the Last Glacial Maximum (LGM), mid-Holocene (MH), present day and 2070s) using the CCSM4 atmospheric circulation model. In the Maxent model, 30% of the distribution data were selected as the test dataset, and the rest of the data were selected as the training dataset. The maximum number of iterations was set to 5000 to ensure that the model had enough time to converge. Finally, the SDM results were visualized, and the suitable habitat areas were inferred using ArcGIS v10.7. The present-day data for the 19 environmental factors at each P. meyeri and P. mongolica distribution point were extracted using ArcGIS v10.7 (Table S2). The Pearson correlation coefficients for the bioclimatic variables were calculated using the ‘cor’ function in R v4.3.1, retaining only one variable for pairs with |r| > 0.7. Additionally, the variance inflation factor (VIF) for each variable was calculated with the vegan v2.6.4 package in R v4.3.1 to evaluate multicollinearity, ensuring that the retained variables had a VIF < 10. Finally, on the basis of the Pearson correlation coefficients and the VIFs, four variables (BIO01: annual mean temperature; BIO03: isothermality; BIO12: annual precipitation, and BIO15: precipitation seasonality) were used for the combined P. meyeri and P. mongolica analysis. The genetic distances (FST/(1-FST)) for pairs of species were calculated based on the SNPs (Dataset I) using VCFtools v0.1.11. Environmental distances, representing isolation by environment (IBE), were computed using Euclidean distances with the R v4.3.1 package vegan v2.6.4. Geographical distances, representing isolation by distance (IBD), were calculated using Geodesic with the R v4.3.1 package geodist v0.08. To disentangle the effects of IBD and IBE, the partial Mantel test with the R package vegan v2.6.4 was used to quantify IBD while controlling for environmental effects and to quantify IBE while controlling for geographical effects. The significance of the partial Mantel tests was assessed through 999 permutations. An RDA of genetic variation (SNPs, Dataset I) was conducted using the vegan v2.6.4 package in R v4.3.1 to assess the impact of environmental factors, with affected SNPs identified based on a p value threshold (p < 0.05). These SNPs were subsequently mapped onto the reference genome of P. abies to identify genes influenced by environmental factors. The candidate genes were subjected to Gene Ontology (GO) analysis via the OMICSHARE cloud platform (http://www.omicshare.com/tools/). Additionally, BLASTX was used to compare these genes with the Arabidopsis thaliana proteome to elucidate protein function information, with an E-value threshold set at 1e-5. To detect regions under significant selective sweeps in P. meyeri and P. mongolica, we used P. koraiensis as a background group based on the phylogenetic tree results and compared P. meyeri and P. mongolica separately with this background group to identify regions under selection. Additionally, we compared the selected regions between P. mongolica and P. meyeri. We selected windows with significantly high π ratios (top 5%) and significantly high FST values (top 5%) as regions showing significant signals of selective sweeps. This was accomplished using VCFtools v0.1.11 to compute π and FST based on Dataset I with a sliding window approach (10,000-kb windows sliding in 1000-kb steps). Additionally, we conducted Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis on the genes within these selected regions using the OMICSHARE cloud platform (http://www.omicshare.com/tools/) with an E-value threshold set at 1e-5. This enabled us to understand the biological functions of the selected genes. A total of 223 Picea samples were sequenced using GBS, yielding 1182.66 Gb of raw data. After filtering, 1138.58 Gb of clean data were obtained, with an average of 5.11 Gb of clean data per sample. The sequencing quality was high, with Q20 > 97.27% and Q30 > 92.71%. A total of 5,716,899 raw SNPs were called, and filtering yielded 1,290,066 high-quality SNPs. The ML phylogenetic tree (Fig. 3) showed that by using P. pungens and P. likiangensis as outgroups, the 15 populations could be divided into three clades. The first clade consisted of two populations of P. koraiensis, the second clade comprised nine populations of P. meyeri, and the third clade encompassed four populations of P. mongolica. Among the P. meyeri populations, three collected from Hebei Province were distinguishable from six collected from Shanxi Province. Notably, the four populations from the Luya Mountain area in **nzhou, Shanxi, were closely related. Among the P. mongolica populations, the Baiyinaobao population differed from the other three groups. Furthermore, we evaluated the population structure of P. koraiensis, P. meyeri, and P. mongolica under different K values (Fig. 3), and the CV error (Fig. S1) decreased when K ranged from 2 to 6. At K = 2, P. koraiensis and P. meyeri formed distinct genetic components, with P. mongolica being composed of two genetic components. As K increased, P. mongolica differentiated from the geographic populations of P. meyeri earlier to form an independent genetic component, suggesting significant differences between P. mongolica and the various P. meyeri geographic populations. At K = 4, two P. meyeri populations near the P. mongolica distribution area differentiated earlier to form an independent genetic component. At K = 6, the four populations from the Luya Mountain area clustered into one genetic component, consistent with the evolutionary tree results. To understand the population structure characteristics, we also used principal component analysis (PCA) to reveal the relationships among populations. As shown in Fig. 4, P. koraiensis, P. meyeri, and P. mongolica were distinguishable, and P. mongolica was more closely related to P. meyeri, with the classification results being consistent with the evolutionary tree and structural analysis findings. ML phylogenetic tree and structure histogram based on SNP markers. Pun: P. pungens; Lik: P. likiangensis. The remaining species abbreviations are the same as those in Fig. 1 Principal component analysis of Picea populations. The species abbreviations are the same as those in Fig. 1 Overall, based on the π values calculated from the whole-genome SNPs (Table 1), the nucleotide diversity of P. mongolica was greater than that of P. meyeri and P. koraiensis. The P. mongolica population Mon1 had the highest π value of 1.5009 × 10−5, followed by the Mon4 population, with a π value of 1.4968 × 10−5. The lowest π value was found for the P. koraiensis population Kor1, at 1.3156 × 10−5. The observed heterozygosity (Ho) of the P. koraiensis, P. meyeri and P. mongolica populations were all lower than the expected heterozygosity (He). The Ho and He of the P. mongolica populations Mon1 and Mon4 were greater than those of the other populations. The lowest He of 0.1810 was observed for the P. koraiensis population Kor1, and the P. mongolica population Mon2 had the lowest Ho of 0.1422. The Tajima’s D values for all 15 populations of the three species were greater than 0, suggesting that these species had rare alleles at low frequencies and likely underwent population contraction. Overall, the Tajima’s D value for P. mongolica was lower than that for P. meyeri and P. koraiensis. The highest Tajima’s D value was found for the P. koraiensis population Kor2, at 0.5857, followed by the Kor1 population, at 0.5420. The P. mongolica population Mon4 had the lowest value, at 0.3945. As indicated in Fig. 5, the interspecific differences between P. koraiensis and the other two species, P. meyeri and P. mongolica, were significant, indicating a greater degree of differentiation for P. koraiensis. The intraspecific differences within P. meyeri and P. mongolica were smaller than the interspecific differences between them, which was consistent with the results of the phylogenetic and population structure analyses. Interspecific genetic differentiation of P. koraiensis, P. meyeri and P. mongolica. The species abbreviations are the same as those in Fig. 1 We first employed the ABBA–BABA method to analyze gene flow among three species: P. koraiensis, P. meyeri, and P. mongolica. A total of 3794 alleles were shared between P1 and P2, and 3652 alleles were shared between P1 and P3, with a D value of 0.019 and a Z score of 20.16, indicating significant asymmetrical gene flow between P. koraiensis and the two sister groups; that is, there was significant gene flow between P. koraiensis and P. meyeri (Fig. 6). To clarify the direction of gene flow among these three species, we utilized TreeMix analysis to construct a ML tree (Fig. 7). The optimal number of migration edges was eight (Fig. S2), suggesting eight gene flow events among the three species. Historically, there was heavy gene flow from P. koraiensis to P. meyeri but no significant gene flow from P. koraiensis to P. mongolica, consistent with the ABBA–BABA results. Additionally, we revealed a history of strong gene flow from P. meyeri to P. mongolica, three gene flow events from P. mongolica to P. meyeri, and three intraspecific gene flow episodes between P. meyeri populations, indicating frequent gene flow within P. meyeri populations and between P. meyeri and P. mongolica. ABBA–BABA test tree models for P. koraiensis, P. meyeri and P. mongolica. Pun: P. pungens; Lik: P. likiangensis; Kor: P. koraiensis; Mey: P. meyeri; Mon: P. mongolica Gene flow among P. koraiensis, P. meyeri and P. mongolica. The species abbreviations are the same as those in Fig. 1 As illustrated in Fig. 8, the effective population size (Ne) of P. meyeri began to gradually decline beginning in the early Pleistocene (approximately 2 Ma), followed by a period of stability. A sharp decline occurred during the middle Pleistocene (0.8 Ma), indicating a population bottleneck, after which the effective population size stabilized. Ne continued to decrease gradually from 0.38 Ma to 0.2 Ma, stabilized until 0.1 Ma, and once again started to decrease rapidly. For P. mongolica, Ne experienced a sharp decline during the early Pleistocene (2.3–2.0 Ma), leading to a population bottleneck, after which it began to expand at approximately 1.2 Ma, reaching a minor peak before stabilizing. Subsequently, rapid decreases occurred at 0.45 Ma and 0.09 Ma (Fig. 8). Overall, the effective population sizes of P. meyeri and P. mongolica gradually decreased. Demographic history of P. meyeri (A) and P. mongolica (B). Dark gray lines: 75% confidence intervals of the inferences. Light gray lines: 95% confidence intervals of the inferences The area under the curve (AUC) values of the Maxent model for both P. meyeri and P. mongolica were greater than 0.9 (Fig. S3), indicating excellent model accuracy. Jackknife tests and the percentage contribution results (Fig. S4) showed that BIO02 (mean diurnal range), BIO04 (temperature seasonality), BIO10 (mean temperature of the warmest quarter), BIO13 (precipitation of the wettest month), BIO15 (precipitation seasonality), and BIO17 (precipitation of the driest quarter) were key environmental factors for predicting the geographic distribution of P. meyeri. For P. mongolica, the best environmental predictors were BIO03 (isothermality), BIO04, BIO09 (mean temperature of the driest quarter), BIO15, and BIO17. Thus, seasonal variations in temperature and precipitation, as well as the minimum amount of precipitation, had significant impacts on the distributions of P. meyeri and P. mongolica. The current potential habitat of P. meyeri is located mainly in the Lüliang, Taihang, and Yanshan Mountains, covering provinces and cities such as Shanxi, Hebei, and Bei**g. The total area of the potential habitat is approximately 5.02 × 105 km2, with 2.85 × 104 km2 being highly suitable and 1.08 × 105 km2 being moderately suitable. The current potential habitat of P. mongolica is located primarily at the eastern edge of the Hunshandake Desert, which includes parts of Inner Mongolia, with a total area of approximately 3.67 × 104 km2, including 1.39 × 103 km2 of highly suitable land and 1.51 × 104 km2 of moderately suitable land. Under historical climatic conditions, the potential habitat areas of P. meyeri and P. mongolica continuously decreased from the LGM to the MH but significantly expanded from the MH to the present. The area of highly suitable habitat for P. meyeri and P. mongolica expanded from the LGM through the MH to the present. The distribution ranges of both species showed trends of moving northeastward from the LGM to the present. Simultaneously, the potential geographical distributions of the two Picea species under two emission scenarios for the future (2070s) were simulated. Under the RCP2.6 emission scenario, the total suitable area for P. meyeri would be 3.26 × 105 km2, with a highly suitable area of 1.34 × 104 km2, representing decreases of 35.18% and 52.80%, respectively, from the current potential distribution area. The total suitable area for P. mongolica would be 3.00 × 103 km2, a decrease of 91.73% from the current potential distribution, and the highly suitable area would disappear. Under the RCP8.5 emission scenario, the total suitable area for P. meyeri would be 1.68 × 105 km2, with a highly suitable area of 0.07 × 104 km2, representing drastic decreases of 66.53% and 97.45%, respectively, from the current potential distribution, while the suitable habitat for P. mongolica would disappear (Fig. 9). Prediction of potentially suitable habitats for P. meyeri (A) and P. mongolica (B) As illustrated in Fig. 10, genetic distance was strongly associated with environmental distance for P. meyeri and P. mongolica combined, indicating local adaptation to the environment within populations of these two spruce species. Genetic distance was also significantly associated with environmental distance based on four environmental variables. The results for IBD and IBE confirm that both geographic and environmental distances strongly influence genetic distance. Among the whole-genome SNPs distributed across 13 populations of P. meyeri and P. mongolica, 96,543 were associated with the environmental factors BIO01, BIO03, BIO12, and BIO15 (Table S3). Figure 11 shows that the first three axes of the RDA explained 39.33%, 28.46%, and 18.51% of the variation, respectively. BIO15 contributed most significantly to the genetic variation in the four populations of P. mongolica and the Mey1 population. BIO3 explained most of the genetic variation in the Mey2, Mey3, Mey4, Mey5, and Mey7 populations. BIO1 made the most significant contribution to the genetic variation in Mey8 and Mey9, while BIO03 and BIO12 explained most of the genetic variation in Mey6. The GO enrichment analysis results (Fig. S5) suggested that the potential candidate genes related to environmental variables are involved mainly in biological processes such as the response to water deprivation, response to abiotic stimuli, and response to hormones. The main molecular functions of these genes included carbohydrate derivative binding, phospholipase activity, and hydrolase activity. Using BLASTX, candidate genes were compared against the Arabidopsis protein database, revealing many proteins related to environmental adaptability (Table S4), such as proteins related to drought (e.g., PUB22/23, BAM1, and SUS3), abiotic stress (e.g., CAT2, Sect. 14, and ERD4), and disease resistance (e.g., RDR1, TAO1, and SGT1b). Partial Mantel tests of genetic distance against (A) geographical distance and (B) environmental distance RDA of genetic variation and environmental variables. The species abbreviations are the same as those in Fig. 1 A total of 167 genes under selection were identified across these three comparisons. Specifically, 65 common genes under selection were identified between P. meyeri and the reference group (Table S5), 72 genes under selection were common between P. mongolica and the reference group (Table S6), and 30 genes under selection were common between P. meyeri and P. mongolica (Table S7). Figure S6 clearly shows that the selective sweep areas between P. meyeri and the reference group are defined by πkor/πmey ≥ 2.89 and FST ≥ 0.34. The GO enrichment analysis (Table S8) revealed that the selected genes were annotated with 202 terms, among which 145, 30, and 27 belonged to the biological process (BP), cellular component (CC), and molecular function (MF) subgroups, respectively. The MFs were mainly associated with protein heterodimerization activity, hydrolase activity acting on ether bonds, adenosylhomocysteinase activity, etc. (Fig. 12). The selective sweep areas between P. mongolica and the reference group were defined by πkor/πmon ≥ 2.14 and FST ≥ 0.33 (Fig. S6). Based on the GO enrichment analysis (Table S9), selected genes were annotated with 290 terms, with 210, 33 and 47 terms in the BP, CC, and MF subgroups, respectively. The MF terms were primarily those associated with GTPase binding, adenosylhomocysteinase activity, hydrolase activity acting on ether bonds, etc. (Fig. 12). The KEGG enrichment analysis (Fig. 13) indicated that the genes were involved mainly in pathways related to nucleocytoplasmic transport, cysteine and methionine metabolism, and C5-branched dibasic acid metabolism. The selective sweep areas between P. mongolica and P. meyeri were defined by πmey/πmon ≥ 1.56 and FST ≥ 0.14 (Fig. S6). The GO enrichment analysis (Table S10) revealed that the selected genes were annotated with 166 terms, among which 108, 44, and 14 belonged to the BP, CC, and MF subgroups, respectively. The MFs were mainly those related to ATP binding, structural constituents of ribosomes, importin-alpha family protein binding, etc. (Fig. 12). The KEGG enrichment analysis (Fig. 13) revealed that the genes were involved in pathways such as neomycin, kanamycin and gentamicin biosynthesis and ribosomes. GO analysis of genes experiencing selective sweeps KEGG pathway analysis of genes experiencing selective sweeps P. meyeri and P. mongolica are unique, ecologically important spruce tree species native to China. However, the taxonomic status of P. mongolica has been controversial. Our study revealed that these two species form independent branches in a phylogenetic tree and that P. mongolica formed independent genetic clusters earlier than the geographical populations of P. meyeri. It also formed a distinct cluster separate from P. meyeri and P. koraiensis in the PCA. Furthermore, the interspecies FST between P. mongolica and P. meyeri was greater than the intraspecific FST of P. meyeri, suggesting that P. mongolica is an independent taxonomic species at the genomic level. This finding is consistent with the results of other studies based on RAPD and ISSR molecular markers and chloroplast genes [17, 18, 20] but differs from the results of morphological comparisons [11,12,13]. This discrepancy may be due to weak reproductive isolation within the genus Picea and frequent gene flow between species leading to phenotypic convergence [40]. Moreover, TreeMix analysis revealed four gene flow events between P. meyeri and P. mongolica. The ABBA–BABA results revealed significantly more gene flow between P. koraiensis and P. meyeri than between P. koraiensis and P. mongolica. This finding is consistent with the greater interspecies FST between P. mongolica and P. koraiensis than between P. mongolica and P. meyeri, indicating that P. mongolica and P. meyeri are closely related. Through structural analysis, we found that when K = 2, which represents the optimal grou**, P. mongolica consists of approximately 50% genetic components from P. koraiensis and 50% from P. meyeri. This suggests that its ancestral type is a hybrid of P. meyeri and P. koraiensis, corroborating the inference of a reticulate evolutionary history for spruce species [40]. The P. mongolica genetic clusters were mixed with the genetic clusters of three P. meyeri populations in Hebei. Moreover, the P. meyeri populations in Hebei Province exhibited a closer genetic relationship with P. mongolica than did the P. meyeri populations in Shanxi Province. This suggests that the Hebei P. meyeri populations represent transitional populations, consistent with their geographical distribution. The uplift of the Mongolian Plateau at approximately 5 ± 3 Ma created geographical isolation, leading to the differentiation of P. meyeri from P. mongolica [41]. P. mongolica evolved in a relatively isolated geographical environment, while P. meyeri migrated southward during the late Pliocene and early Pleistocene when the climate became drastically colder [42], spreading from Hebei to Shanxi. Long-term evolutionary processes and environmental influences shape and preserve genetic variations within populations [43]. Despite the clear geographical differences between P. meyeri and P. mongolica, frequent gene flow still occurs between them, a phenomenon that is also often observed in other isolated plant lineages [44,45,46]. Speciation with gene flow may be common in nature [47]. Spatial heterogeneity weakens gene flow, and the divergent selection pressures arising from heterogeneous habitats are a primary driving factor for adaptive speciation [45, 48]. However, during the process of adaptive speciation, gene flow aggregates genetic variations from different populations into a single population, thereby enhancing the genetic variation mediated by selection, which accelerates species diversification [49,50,51]. Recent research has indicated that moderate gene flow may facilitate adaptive differentiation, primarily because gene flow can reduce the linkage between genetic loci through recombination and segregation, increasing genetic differentiation and thereby promoting speciation [51, 52]. Thus, P. mongolica may have originated as a local variety of P. meyeri due to geographical isolation and then gradually evolved into an independent species of the Picea genus, adapting to sandy environments. However, studies based on chloroplast genes suggest that P. mongolica is an ancestor of P. meyeri and P. koraiensis [20], possibly due to recent radiating differentiation, reticulate evolution, and interspecies plastid recombination in the Picea genus [53]. Despite the limited distribution range of P. mongolica, this species exhibits greater nucleotide diversity than both P. meyeri and P. koraiensis, consistent with the findings of previous chloroplast gene-based studies [20]. This high nucleotide diversity enables P. mongolica to persist within its narrow distribution range, enhancing its adaptability to sandy environments. Due to their adjacent distributions, P. meyeri and P. mongolica experienced multiple gene flow events during their differentiation processes [40, 54]. However, due to differences in their ecological environments, they might be strongly influenced by specific environmental factors during evolution, leading to population differentiation at key loci [55, 56]. Subsequently, advantageous mutations may have been preserved through selection, leading to their proliferation and expansion, thereby continuously promoting genetic differentiation within populations, a phenomenon commonly observed in closely related species with adjacent distributions but different ecological niches [57, 58]. Temperature and precipitation are crucial factors affecting species distributions and growth. In this study, IBE and RDA were used to determine the relationships between environmental factors and genetic variation. BIO01, BIO3, BIO12, and BIO15 are important factors explaining the local environmental adaptation of the two Picea species. P. mongolica occupies the eastern edge of the Hunshandake sandy land, with concentrated stands on both the leeward and windward slopes of the Baiyinaobao sandy land, making it one of the preferred tree species for afforestation in China’s sandy lands [59, 60]. Studies on the P. mongolica growth conditions and ecological requirements in sandy lands have shown that P. mongolica growth in June accounts for approximately 65% of its annual growth, with the water and thermal conditions in June determining its annual height increase, making seasonal precipitation particularly important for its growth [61]. Conversely, P. meyeri is distributed in areas such as Shanxi, Hebei, and parts of Inner Mongolia, where the temperature, rainfall, and humidity are greater than those in the habitats of P. mongolica; thus, temperature and annual precipitation significantly impact its distribution. Among the candidate genes related to climate factors, genes associated with plant drought resistance were detected. BAM1, SUS3, and CIPK23 can regulate stomatal opening in response to water stress [62,63,64], while PUB22 and PUB23 coordinate the control of drought signaling pathways [65]. CAT2, Sect. 14, and ERD4 enhance plant salt stress tolerance [66,67,68], and EBF1 and MPK6 enhance plant cold tolerance [69, 70]. Plants often adapt to abiotic conditions such as climate through positive selection, which leaves traces of selective sweeps on genes linked to those that are selected [2]. Selective sweeps usually reduce genetic diversity within a population but enhance plant environmental adaptability, thereby promoting local adaptation [71]. In the present study, P. koraiensis was used as a reference, and genes showing evidence of selective sweeps in P. meyeri and P. mongolica were related mainly to plant growth and stress resistance. Genes such as SAHH, SPX1, and ERF042 were found to be under selection in both species. The SAHH gene plays a key role in secondary cell wall biosynthesis in trees [72]. P. koraiensis, which is distributed mainly in Northeast China, experiences low winter temperatures, and lignin synthesis in secondary cell walls helps plants resist cold conditions. The SPX1 gene is closely related to phosphorus homeostasis during plant growth under phosphorus deficiency [73]. P. mongolica mainly inhabits sandy habitats where phosphorus is relatively scarce, while P. meyeri is mainly distributed in brown forest soil areas with high rainfall and humidity, where phosphorus is easily leached by percolation. ERF genes are involved in plant development and stress response regulation. Studies have shown that ERF042 is related to flower development in angiosperms [74]. The environmental differences between P. meyeri and P. mongolica have led to differences in their phenological traits. In P. meyeri, genes related to plant temperature adaptation, such as HSP70-6 [75], and those related to pollen development and, in angiosperms, flowering, such as AMS, MYB2, and HD3A [76,77,78], were also found, reflecting the phenological differences in their distribution areas. In P. mongolica, genes related to root growth, such as ACT2 [79]; drought stress, such as KPNB1 [80]; and vascular development, such as ERF018 [81], were found to aid in adaptation to arid environments in sandy areas. With P. meyeri as the reference, genes related to stomatal regulation, such as KIN10A [82]; drought or cold stress, such as SRP, RGGA, and HXK2 [83,84,85]; and vascular plant seed germination and fruit ripening, such as PME [86], were found in P. mongolica, indicating that these selectively swept genes enable better adaptation to sandy environments and contributed to its evolution into an independent species within the genus Picea. Other researchers have also identified genes related to stomatal regulation, flowering (in angiosperms), and stress resistance among the genes showing evidence of selective sweeps in P. asperata, P. crassifolia, P. wilsonii, and P. koraiensis [58, 87, 88]. Throughout history, the habitat of the genus Picea has undergone significant changes due to geological events and climate fluctuations [89], resulting in new selection pressures. The process of selective sweeps, which rapidly fixes advantageous genes, has not only enhanced the adaptation of Picea species to their environments but has also been a driving force in the diversification of species within the genus [71]. However, the cloning and functional validation of these selected genes remain to be performed. The stairway plot results indicate that the Ne values of P. meyeri and P. mongolica are overall in a state of continuous decline. The Ne of P. meyeri sharply decreased toward the end of the Calabrian period of the Pleistocene due to colder and drier climate conditions, and it likely decreased during the Dali glaciation due to lower temperatures. The Ne of P. mongolica dramatically decreased during the mid-Pleistocene climate transition, which was attributed to the significant increase in aridification [90]. After the species adapted to the arid climate, its Ne increased, followed by rapid decreases during the early and late phases of the LGM. The SDM results revealed that the total P. meyeri and P. mongolica suitable habitat continuously decreased from the LGM to the MH and then expanded from the MH to the present. However, Ne has been continuously decreasing, which may be due to the substantial reduction in Ne caused by earlier environmental changes. In recent years, human activities have led to continuous reductions in forest resources, especially for P. mongolica, which inhabits the ecotone and transition zone of the Hunshandake sandy land, where the pressures from pests and diseases have sharply increased [91, 92]. SDM simulations under future climate change scenarios revealed decreases in the suitable distribution areas for both species, with greater reductions in highly suitable areas and accelerated decreases with increasing greenhouse gas emissions and concentrations. Particularly under high-emission scenarios, P. mongolica could lose almost all of its suitable habitat. Notably, P. mongolica possesses high genetic diversity and has great potential for adapting to climate change, making it a valuable breeding material. Therefore, P. mongolica should be considered an independent species, and conservation measures should be implemented to prevent its extinction. Existing studies indicate that purposeful and conscientious human intervention can promote natural regeneration and the recovery of genetic diversity in natural spruce communities in sandy lands [93]. For P. mongolica populations facing the risk of extinction, it is necessary to strengthen the management of water conditions in sandy natural forests to meet their natural regeneration needs. Genetic resource surveys and the collection and management of P. meyeri and P. mongolica should be conducted, and on this basis, in situ and ex situ conservation areas should be established. Additionally, comprehensive germplasm resource gene banks need to be established to prevent the loss of genetic diversity. The GBS of 188 individuals from 13 P. meyeri and P. mongolica populations revealed genetic differences between the two species. Genomically, although the ancestor of P. mongolica may have been a hybrid, P. mongolica can be considered an independent species. There have been multiple instances of gene flow between the two species. The Ne of P. meyeri decreased from the Early Pleistocene to the present, while that of P. mongolica slightly decreased after an initial decline in the Early Pleistocene, followed by a continuous decrease. The total suitable habitat area for both species showed a trend of initially decreasing and then increasing from the previous geological era to the present but is projected to decrease under future climate scenarios. Local adaptation may have facilitated the differentiation of these two species, and selective sweeps revealed candidate genes mainly associated with plant organ development and resistance. The results deepen our understanding of the reticulate speciation mechanisms within the Picea genus. Future research could integrate plant phenotypes to explore the mechanisms of interaction between gene function, plant phenotype, and environmental adaptability.Materials and methods
Plant materials


DNA extraction and sequencing and SNP calling
Phylogenetic and population structural analyses
Gene flow
Population dynamics analysis
Species distribution model (SDM)
Environmental association analysis
Selection of environmental variables
Isolation by environment (IBE) and isolation by distance (IBD)
Redundancy analysis (RDA)
Selective sweep analyses
Results
Sequencing quality
Phylogenetic and population structure analyses


Population diversity and genetic divergence

Gene flow


Historical population dynamics of P. Meyeri and P. Mongolica

Changes in potential habitats

Environmental adaptability and genetic variation


Genes experiencing selective sweeps in P. Meyeri and P. Mongolica


Discussion
Species differentiation and gene flow characteristics
Signatures of environmental adaptation of the two Picea species
Population dynamics and conservation management of two Picea species
Conclusion
Availability of data and materials
The raw sequencing data generated from this study have been deposited in NCBI SRA (https://www.ncbi.nlm.nih.gov/sra) under the accession number PRJNA876367.
Abbreviations
- GBS:
-
Genoty**-by-sequencing
- Ne:
-
Effective population size
- LGM:
-
Last Glacial Maximum
- MH:
-
Mid-Holocene
- Pun:
-
P. pungens
- Lik:
-
P. likiangensis
- Kor1, Kor2:
-
P. koraiensis from Mengke mountain and Linjiang, respectively
- Mey1, Mey2, Mey3, Mey4, Mey5, Mey6, Mey7, Mey8, Mey9:
-
P. meyeri from Saihanba, Kelan, Wuzhai, Shenchi, Ningwu, Jiaocheng, Wutai mountain, **aoWutai mountain, and Wuling mountain, respectively
- Mon1, Mon2, Mon3, Mon4:
-
P. mongolica from Baiyinaobao, Dajuzi, Huanggangliang, and Huamugou, respectively
References
Leimu R, Fischer M. A meta-analysis of local adaptation in plants. PLoS One. 2008;3:e4010.
Wei K, Silva-Arias GA, Tellier A. Selective sweeps linked to the colonization of novel habitats and climatic changes in a wild tomato species. New Phytol. 2023;237:1908–21.
Sobel JM, Chen GF, Watt LR, Schemske DW. The biology of speciation. Evolution. 2010;64:295–315.
Wright KM, Lloyd D, Lowry DB, Macnair MR, Willis JH. Indirect evolution of hybrid lethality due to linkage with selected locus in Mimulus guttatus. PLoS Biol. 2013;11:e1001497.
Hoffmann AA, Sgrò CM. Climate change and evolutionary adaptation. Nature. 2011;470:479–85.
Liu J. The integrative species concept and species on the speciation way. Biodivers Sci. 2016;24:1004–8.
Wu SD, Wang Y, Wang ZF, Shrestha N, Liu J. Species divergence with gene flow and hybrid speciation on the Qinghai–Tibet Plateau. New Phytol. 2022;234:392–404.
Farjón A. A handbook of the world’s conifers. Second, revised edition. Leiden, Netherlands: Brill; 2017.
Fu LG, Li N, Elias TS. Picea. Flora of China. Bei**g, China: Science; 1999. pp. 25–32.
Shen TT, Ran JH, Wang XQ. Phylogenomics disentangles the evolutionary history of spruces (Picea) in the Qinghai-Tibetan Plateau: implications for the design of population genetic studies and species delimitation of conifers. Mol Phylogenet Evol. 2019;141:106612.
Zheng W. Tree Flora of China. Bei**g, China: China Forestry Publishing House; 1983.
Xu W. Picea Meyeri and its spruce forest in the Nei Monggol sands. Chin J Plant Ecol. 1983;7:1–7.
Wu H. A new variety of Picea dietr. Bull Bot Res. 1986;6:153–5.
Li C, Lan D, Zhou S, Zhao X, **ng J. Research on the taxonomy of species of Picea in Baiyinaobao sandy land of Inner Mongolia. J Arid Land Resour Environ. 2008;22:164–9.
Xu W, Li W, Zheng Y. A study on taxonomy of Picea Mongolica in Inner Mongolia. Bull Bot Res. 1994;14:59–69.
Zou CJ, Han SJ, Xu W, duo SB. Formation, distribution, and nature of Picea Mongolica in China. J For Res. 2001;12:187–91.
Cai P, Wan T, Zhang H, Yi W, Li F, Meng X, et al. RAPD analysis on genetic diversity of Picea Mongolica (H.Q.Wu.) W.D.Xu and its close relative species. J Agric Sci Technol. 2009;11:102–10.
Zheng S, Bai Y, He Y, Tian Y. The interspecific genetic diversity of Picea Mongolica and its relative species. J Fujian Sci Technol. 2015;42:24–8.
Bai Y, Peng P, Wang J, Yi R, Duan G, Lin T. Comparative FISH karyotype analysis of Picea Mongolica and its closest relatives. Mol Plant Breed. 2016;14:3548–53.
Di H, Ma JJ, He KK, Han F, Li Y, Niu S. Phylogenetic relationship of Picea mongolica with other Picea species in the same area based on chloroplast gene variations. J Res. 2021;32:297–305.
Duan G, Bai Y, Yi R, Bai S. GISH analysis of Picea Mongolica and related species. Acta Bot Boreal-Occident Sin. 2018;38:0225–31.
Tel-zur N, Abbo S, Myslabodski D, Mizrahi Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae). Plant Mol Biol Rep. 1999;17:249–54.
Poland JA, Brown PJ, Sorrells ME, Jannink J-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genoty**-by-sequencing approach. PLoS One. 2012;7:e32253.
Poland J, Endelman J, Dawson J, Rutkoski J, Wu S, Manes Y, et al. Genomic selection in wheat breeding using genoty**-by-sequencing. Plant Genome. 2012;5:103–13.
Chen SF, Zhou YQ, Chen YR, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967.
Malinsky M, Matschiner M, Svardal H. Dsuite - fast D ‐statistics and related admixture evidence from VCF files. Mol Ecol Resour. 2021;21:584–95.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Liu XM, Fu YX. Stairway plot 2: demographic history inference with folded SNP frequency spectra. Genome Biol. 2020;21:280.
Bodare S, Stocks M, Yang J, Lascoux M. Origin and demographic history of the endemic Taiwan spruce (Picea Morrisonicola). Ecol Evol. 2013;3:3320–33.
Tsuda Y, Chen J, Stocks M, Källman T, Sønstebø JH, Parducci L, et al. The extent and meaning of hybridization and introgression between siberian spruce (Picea Obovata) and Norway spruce (Picea abies): cryptic refugia as step** stones to the west? Mol Ecol. 2016;25:2773–89.
Wuyun T. Phylogeography of Picea mongolica and its related species. Hohhot: Inner Mongolia Agricultural University; 2020.
Feng S, Ru D, Sun Y, Mao K, Milne R, Liu J. Trans-lineage polymorphism and nonbifurcating diversification of the genus Picea. New Phytol. 2019;222:576–87.
Wang Y. Phylogeography of Picea koraiensis and its closely related species based on simplified genome. PhD diss. Bei**g: Chinese Academy of Forestry; 2023.
Sosdian S, Rosenthal Y. Deep-sea temperature and ice volume changes across the Pliocene-Pleistocene climate transitions. Science. 2009;325:306–10.
Ru DF, Sun YS, Wang DL, Chen Y, Wang T, Hu Q, et al. Population genomic analysis reveals that homoploid hybrid speciation can be a lengthy process. Mol Ecol. 2018;27:4875–87.
Zhang D, Hao GQ, Guo XY, Hu Q, Liu J. Genomic insight into sky island species diversification in a mountainous biodiversity hotspot. J Sytematics Evol. 2019;57:633–45.
Ma YZ, Wang J, Hu QJ, Li JL, Sun YS, Zhang L, et al. Ancient introgression drives adaptation to cooler and drier mountain habitats in a cypress species complex. Commun Biol. 2019;2:213.
Li JL, Zhong LL, Wang J, Ma T, Mao K, Zhang L. Genomic insights into speciation history and local adaptation of an alpine aspen in the Qinghai–Tibet Plateau and adjacent highlands. J Syst Evol. 2021;59:1220–31.
Li Z, Liu Z, Wang M, Qian Z, Zhao P, Zhu J, et al. A review on studies of speciation in the presence of gene flow: evolution of reproductive isolation. Biodivers Sci. 2014;22:88–96.
Elias M, Faria R, Gompert Z, Hendry A. Factors influencing progress toward ecological speciation. Int J Ecol. 2012;2012:1–7.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Nosil P, Gompert Z, Farkas TE, Comeault AA, Feder JL, Buerkle CA, et al. Genomic consequences of multiple speciation processes in a stick insect. Proc R Soc B. 2012;279:5058–65.
Flaxman SM, Feder JL, Nosil P. Genetic hitchhiking and the dynamic buildup of genomic divergence during speciation with gene flow: special section. Evolution. 2013;67:2577–91.
Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Mol Ecol. 2009;18:375–402.
Sullivan AR, Schiffthaler B, Thompson SL, Street NR, Wang X-R. Interspecific plastome recombination reflects ancient reticulate evolution in Picea (Pinaceae). Mol Biol Evol. 2017;34:1689–701.
Barton NH. What role does natural selection play in speciation? Phil Trans R Soc B. 2010;365:1825–40.
Savolainen O, Lascoux M, Merilä J. Ecological genomics of local adaptation. Nat Rev Genet. 2013;14:807–20.
Tiffin P, Ross-Ibarra J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol Evol. 2014;29:673–80.
Hu HY, Yang YZ, Li A, Zheng Z, Zhang J, Liu J. Genomic divergence of stellera chamaejasme through local selection across the Qinghai–Tibet plateau and northern China. Mol Ecol. 2022;31:4782–96.
Feng S, Wan W, Li Y, Wang D, Ren G, Ma T, et al. Transcriptome-based analyses of adaptive divergence between two closely related spruce species on the Qinghai–Tibet plateau and adjacent regions. Mol Ecol. 2023;32:476–91.
Wang W, Liang C, Li H, Liu Z. Preliminary study on properties, probable causes and capacity for natural regeneration of Picea Meyeri forest on sandy land in Baiyinxile. J Arid Land Resour Environ. 2000;14:59–64.
Zou CJ, Han SJ, Zhang JH, Wang CR, Zhou Y. Characteristics and dynamics of sandy natural forests in sandy forest-steppe ecotone in the northern area of China. J Res. 2000;11:161–6.
Zheng Y, Xu W. The models of relationship between height growth of Picea Mongolica and site conditions. Chin J Ecol. 1997;16:62–6.
Zanella M, Borghi GL, Pirone C, Thalmann M, Pazmino D, Costa A, et al. β-amylase 1 (BAM1) degrades transitory starch to sustain proline biosynthesis during drought stress. J Exp Bot. 2016;67:1819–26.
Daloso DM, Williams TCR, Antunes WC, Pinheiro DP, Müller C, Loureiro ME, et al. Guard cell-specific upregulation of sucrose synthase 3 reveals that the role of sucrose in stomatal function is primarily energetic. New Phytol. 2016;209:1470–83.
Nieves-Cordones M, Caballero F, Martínez V, Rubio F. Disruption of the Arabidopsis thaliana inward-rectifier K + channel AKT1 improves plant responses to water stress. Plant Cell Physiol. 2012;53:423–32.
Cho SK, Ryu MY, Song C, Kwak JM, Kim WT. Arabidopsis PUB22 and PUB23 are homologous U-box E3 ubiquitin ligases that play combinatory roles in response to drought stress. Plant Cell. 2008;20:1899–914.
Zhang Y, Wang LF, Li TT, Liu W-C. Mutual promotion of LAP2 and CAT2 synergistically regulates plant salt and osmotic stress tolerance. Front Plant Sci. 2021;12:672672.
Qin YX, Zhang B, Wang YN, Su R. Characterization of SEC14 family in wheat and the function of TaSEC14-7B in salt stress tolerance. Plant Physiol Biochem. 2023;202:107926.
Liu YH, Li HY, Shi YS, Song YC, Wang TY, Li Y. A maize early responsive to dehydration gene, ZmERD4, provides enhanced drought and salt tolerance in Arabidopsis. Plant Mol Biol Rep. 2009;27:542–8.
Wu WZ, Yang HB, Shen J, **ng P, Han X, Dong Y, et al. Identification of Brassica rapa BrEBF1 homologs and their characterization in cold signaling. J Plant Physiol. 2023;288:154076.
Saucedo-García M, González-Córdova CD, Ponce-Pineda IG, Cano-Ramírez D, Romero-Colín FM, Arroyo-Pérez EE, et al. Effects of MPK3 and MPK6 kinases on the chloroplast architecture and function induced by cold acclimation in Arabidopsis. Photosynth Res. 2021;149:201–12.
Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D, et al. The role of geography in human adaptation. PLoS Genet. 2009;5:e1000500.
Du QZ, Wang L, Zhou DL, Yang HJ, Gong CR, Pan W, et al. Allelic variation within the S-adenosyl-L-homocysteine hydrolase gene family is associated with wood properties in Chinese white poplar (Populus tomentosa). BMC Genet. 2014;15:S4.
Zhang JY, Zhou X, Xu Y, Yao M, **e F, Gai J, et al. Soybean SPX1 is an important component of the response to phosphate deficiency for phosphorus homeostasis. Plant Sci. 2016;248:82–91.
Jiang Q, Wang Z, Hu GM, Yao XH. Genome-wide identification and characterization of AP2/ERF gene superfamily during flower development in Actinidia Eriantha. BMC Genomics. 2022;23:650.
Zhou C, Yang XB, Yang H, Long GY, Wang Z, ** DC. Effects of abiotic stress on the expression of Hsp70 genes in Sogatella furcifera (Horváth). Cell Stress Chaperones. 2020;25:119–31.
Xu J, Ding ZW, Vizcay-Barrena G, Shi J, Liang W, Yuan Z, et al. ABORTED MICROSPORES acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell. 2014;26:1544–56.
Guo XR, Li LH, Liu XT, Zhang C, Yao X, Xun Z, et al. MYB2 is important for tapetal PCD and pollen development by directly activating protease expression in Arabidopsis. Int J Mol Sci. 2022;23:3563.
Komiya R, Ikegami A, Tamaki S, Yokoi S, Shimamoto K. Hd3a and RFT1 are essential for flowering in rice. Development. 2008;135:767–74.
Vaškebová L, Šamaj J, Ovečka M. Single-point ACT2 gene mutation in the Arabidopsis root hair mutant der1-3 affects overall actin organization, root growth and plant development. Ann Bot. 2017;122:889–901.
Luo YJ, Wang ZJ, Ji HT, Fang H, Wang S, Tian L, et al. An Arabidopsis homolog of importin β1 is required for ABA response and drought tolerance. Plant J. 2013;75:377–89.
Etchells JP, Provost CM, Turner SR. Plant vascular cell division is maintained by an interaction between PXY and ethylene signalling. PLoS Genet. 2012;8:e1002997.
Han C, Liu Y, Shi W, Qiao Y, Wang L, Tian Y, et al. KIN10 promotes stomatal development through stabilization of the SPEECHLESS transcription factor. Nat Commun. 2020;11:4214.
Kim EY, Park KY, Seo YS, Kim WT. Arabidopsis small rubber particle protein homolog SRPs play dual roles as positive factors for tissue growth and development and in drought stress responses. Plant Physiol. 2016;170:2494–510.
Ambrosone A, Batelli G, Nurcato R, Aurilia V, Punzo P, Bangarusamy DK, et al. The arabidopsis RNA-binding protein AtRGGA regulates tolerance to salt and drought stress. Plant Physiol. 2015;168:292–306.
Wang HB, **n H, Guo JY, Gao Y, Liu C, Dai D, et al. Genome-wide screening of hexokinase gene family and functional elucidation of HXK2 response to cold stress in Jatropha curcas. Mol Biol Rep. 2019;46:1649–60.
Li YQ, He HY, He LF. Genome-wide analysis of the pectin methylesterase gene family in potato. Potato Res. 2021;64:1–19.
Liu YF, Qin AL, Wang Y, Nie W, Tan C, An S, et al. Interspecific gene flow and selective sweeps in Picea Wilsonii, P. Neoveitchii and P. Likiangensis. Plants. 2022;11:2993.
Wang Y, Jiang ZP, Qin AL, Wang F, Chang E, Liu Y, et al. Population structure, genetic diversity and candidate genes for the adaptation to environmental stress in Picea Koraiensis. Plants. 2023;12:1266.
Wang Y, Wang J, Jiang Z, Qi X, Jia Z. Study on the phylogeography of Picea. World Res. 2020;33:13–8.
**ong W, Zhu L, Zhang X, Guo L, Ye W, **ong S. Middle Pleistocene transition and its environmental effects revealed by red earth of south China. Quat Sci. 2020;40:1441–52.
Xu W. Basic characteristics, problems and solution ways for spruce forest in sandy land in Baiyinaobao Natural Reserve, Inner Mongolia. Chin J Ecol. 1993;12:39–44.
Liu Z. Study on resources and priority conservation of rare and endangered plants in Inner Mongolia. PhD diss. Hohhot: Inner Mongolia Agricultural University; 2017.
Liu Y, Chen L, Duan W, Bai Y, Zhao Y. Review of the studies on Picea Mongolica forest. Acta Ecol Sin. 2020;40:34–42.
Funding
This work was funded by the National Key Research and Development Program of China (No. 2023YFD2200605-02) and Young Scientists Fund of the National Natural Science Foundation of China (No. 31500540).
Author information
Authors and Affiliations
Contributions
Yifu Liu: Conceptualization, Formal analysis, Investigation, Software, Writing–original draft, Writing–review & editing. Wenfa ** An: Resources. Ermei Chang: Writing–review & editing. Ze** Jiang: Investigation. Junhui Wang: Conceptualization, Writing–review & editing, Data curation, Supervision. Zirui Jia: Conceptualization, Writing–review & editing, Funding acquisition, Data curation, Supervision, Project administration.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., **ao, W., Wang, F. et al. Adaptive divergence, historical population dynamics, and simulation of suitable distributions for Picea Meyeri and P. Mongolica at the whole-genome level. BMC Plant Biol 24, 479 (2024). https://doi.org/10.1186/s12870-024-05166-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05166-6