
Abstract
Background
Hybridization is generally considered an important creative evolutionary force, yet this evolutionary process is still poorly characterized in karst plants. In this study, we focus on natural hybridization in yellow Camellia species, a group of habitat specialists confined to karst/non-karst habitats in southwestern China.
Results
Based on population genome data obtain from double digest restriction-site associated DNA (ddRAD) sequencing, we found evidence for natural hybridization and introgression between C. micrantha and C. flavida, and specifically confirmed their hybrid population, C. “ptilosperma”. Ecophysiological results suggested that extreme hydraulic traits were fixed in C. “ptilosperma”, these being consistent with its distinct ecological niche, which lies outside its parental ranges.
Conclusion
The identified hybridization event is expected to have played a role in generating novel variation during, in which the hybrid population displays different phenological characteristics and novel ecophysiological traits associated with the colonization of a new niche in limestone karst.
Similar content being viewed by others


Background
Natural hybridization is known to occur in numerous lineages, leading to diverse possible evolutionary consequences [1,2,3]. In many case, hybridization simply result in the formation of sterile or maladapted offspring [4, 5]. In other case, hybridization may have contributed to speciation and adaptation by generating genetic variation, functional novelty and even new species [3, 6, 7]. Hybridization occurs more frequently among closely related species and is pervasive in recently diverged lineages, such as those that underwent rapid species radiation [8,9,10]. Such radiation is most likely to happen in mountainous regions, islands and rift-lakes, where alternative resources are more widely accessible [8, 11]. In the context of adaptive radiation, the first step after contact between two established species is the ensuing formation of a hybrid swarm with novel phenotype, which may enable its colonization of novel niches [6, 12]. If hybrids can colonize such niches, become stabilized and eventually lead to speciation [4, 12, 13]. Introgression is also a frequent outcome of hybridization [14]. Adaptive introgression occurs when the transfer genetic material has positive fitness consequences in the recipient species, and is observed between closely related species during the adaptation to new environments [15, 16]. Ecological isolation limits or prevents gene flow and is of itself an important reproductive barrier [17, 18]. Further, the emergence of a new genotype may be directly associated with reproductive isolation, as is the case stark phenological differences arise [19, 20]. Hybridization has now emerged as an important source of adaptation variation, which is quickly introduces much more genetic variation than de novo mutation, and often fuels rapid diversification [2, 15, 21].
Karst landforms are edaphically (soil-related) special terrestrial habitats whose floral composition is unique [22]. In southwestern China, karst terrain with sharp peaks, steep slopes, and deep valleys are conspicuous landscape features, being separated from other outcrops by lowland areas composed of differing soil types [23]. These habitats support high levels of species richness and endemism [24]. Numerous studies have provided valuable insights into karst ecosystems from ecological, physiological, and genetic perspectives, and have greatly improved our understanding of the mechanisms underpinning current species diversity in southwestern China’s karst region. In short, these findings demonstrate that karst forests are influenced by edaphic and hydrological factors related to highly heterogeneous topographies [25, 26]. That body of studies has also revealed that functional trait variation of karst plants influences their species distribution and coexistence [23, 27, 28], and that geographic isolation, genetic drift and selection promotes population differentiation [29,30,31,32,33]. These studies have also reported on the occurrence of whole genome duplication (WGD) events that probably contributed to plants’ adaptation in limestone karst habitats [34]. It is known that hybridization is common in species-rich genera (Begonia) in karst regions [35,36,37]. Karst landforms harbor remarkably high level of plant diversity and given that hybridization is widely acknowledged as a creative force in plant evolution [38], but no study has yet examined hybridization as an aspect of evolution in the diversification of karst plants.
Yellow Camellia species (simply referred to as ‘yellow camellias’), are a group of Camelllia plants whose flowers are yellow. According to the Flora Reipublicae Popularis Sinicae along with subsequent reports of newly described species, there are more than 20 species of yellow camellias that are confined to small areas of southwestern China [39,40,41,42,43,44]. Yellow camellias have very high morphological diversity, including differences in floral color and structure, and leaf morphology. They also display pronounced habitat preferences, with most species associated with limestone substrates and only four species found growing in acidic soils. Only a single known species occurs naturally in both karst and non-karst soils. Such stark differentiation occurs across a range of topographical positions. For example, on a single mountain, C. flavida occupies its karst valley while C. perpatua is restricted to its upper slope near to peak. In addition, several interspecific hybridization events have been inferred among the yellow Camellia plants growing in China [45]. The high richness of species in close proximity to each other, their high degree of habitat specialization, in addition several suspected reticular events together make yellow camellias an excellent model for studying the potential role of hybridization in diversification events in karst regions.
Both C. micrantha and C. flavida are morphologically distinguishable and favor different habitats: C. micrantha grows exclusively in non-karst forests (Figs. 1a, 1b, 1c and 1d), while C. flavida typically inhabits karst depressions (Figs. 1a, 1e, 1h and 1i). Karst soils are typically shallow and are characterized by lower water storage capacity in comparison to the surrounding non-karst soils [46]. Therefore, karst plants frequently incur drought because of low soil water availability, particularly in the dry season [47, 48]. Karst and non-karst species also differ in their hydraulic traits in correspondence with their distribution patterns [28]. Despite this, a plant known as Camellia ptilosperma is suspected to have originated via hybridization between C. micrantha and C. flavida based on the inconsistencies in the phylogenies derived from nuclear and chloroplast genomes [45]. Morphologically similar to C. flavida (Fig. 1f), C. ptilosperma is primarily characterized by long flowering period that distinguish it from other yellow Camellia species, hence why it was initially reported as being a new species [49], albeit later merged with C. flavida [50]. The distribution of this putative hybrid (herein C. “ptilosperma”) is restricted to a very limited area (less than 50 km2, Fig. 1a), in geographic proximity of C. micrantha. It grows on karst slopes, across elevation gradients spanning 230 to 390 m above sea level, characterized by extensive rock outcrops and shallow soils (Fig. 1g). In karst, the soil depth as well as soil distribution continuity generally decrease as the elevation increases [51, 52]. Water availability is the key factor determining the distribution of karst plant species [25, 27], that of C. “ptilosperma” implies these plants probably be more drought-tolerant than species growing in karst valleys/depressions and where the slopes are low. It is speculated that a novel ecological preference might have emerged in C. “ptilosperma”. In theory, tolerance of such habitat in a hybrid population could be conferred by fixation of extreme traits [6, 53, 54]. However, the hybrid origin of C. “ptilosperma” remains untested at population genetic level, and whether C. “ptilosperma” has accordingly achieved niche divergence has not been determined, leaving us with nothing known about its drought tolerance ability.

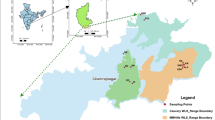
Geographic distribution, morphology, and habitats of the three yellow Camellia taxa studied herein. (a) Sampling locations in southwestern Guangxi, China. (b) Non-karst landscape. (c-d) Morphology and habitat of C. micrantha. (e) Karst landscape. (f-g) Morphology and habitat of C. “ptilosperma”. (h-i) Morphology and habitat of C. flavida. (j) Population structure of the three yellow Camellia taxa based on analyses with the ADMIXTURE program at K = 2, 3, and 4
To this end, here we employed double digest restriction site-associated DNA sequencing (ddRAD-seq) for genomic analysis, and investigated the ecophysiological features associated with habitat adaptation. The specific aims of this study were to: (1) test leading hypotheses about the hybrid origin of C. “ptilosperma”, and (2) to explore whether C. “ptilosperma” has a greater tolerance of specific habitat stresses than its parent species. The obtained data and findings provide for a possible mechanistic basis for lineage diversification in yellow camellias.
Results
SNP genoty**
A total of 2 441 209 808 (mean ± standard deviation [SD], 46 060 562 ± 22 329 937 per individual), 4 004 099 068 (44 001 088 ± 16 071 811 per individual), and 582 841 776 (34 284 810 ± 9 872 024 per individual) reads were obtained from the 53, 91, and 17 individuals of C. micrantha, C. flavida, and C. “ptilosperma”, respectively. The dataset obtained using the STACKS program contained 705 554 SNPs. After filtering, the 4422 SNPs obtained and used for subsequent analyses.
Population structure
Analyses of genetic variations from genome-wide SNP data using the cross-validation (CV) method in ADMIXTURE revealed an optimal value of K = 4 (Fig. S1). For K = 2, the C. “ptilosperma” population showed a high degree of admixed ancestry, with ca. 68% was derived from C. micrantha and ca. 32% derived from C. flavida. Moreover, C. flavida further diverged into different lineages at K = 3 (Fig. 1j). The NG-LLS, NG-MQ, and NG-ND populations showed genetic admixture (Fig. 1j) and the remaining populations showed 100% pure assignment to their respective clusters. For K = 4, most individuals of MZ-SJ population and the C. flavida populations NG-LR and NG-GLA formed a single genetic cluster. Two individuals of the MZ-SJ population still showed a high degree of admixed ancestry with C. micrantha where K = 4 (61% and 57%, Fig. 1j). C. micrantha remained genetically homogeneous across all levels of partitioning, except for the BY-WH population, which featured limited admixture with a cluster comprising the MZ-SJ, NG-LR, and NG-LT populations at K = 4 (Fig. 1j). The ADMIXTURE results also revealed that the NG-LR and NG-GLA populations of C. flavida harbored some degree of introgression from C. micrantha (33%), this being especially distinct at K = 2 and K = 3 (Fig. 1j).
The principal component analysis (PCA) results uncovered four major clusters, which corroborated the result obtained from the ADMIXTURE analysis (Fig. 2a). In this respect, C. micrantha and C. flavida were well differentiated from each other along PC1, which explained 9.31% of the variance. The individuals of C. “ptilosperma” and the NG-LR and NG-GLA populations of C. flavida that showed genetic introgression had intermediate values along PC1, yet were distinguishable along PC2, which explained 4.96% of the variance (Fig. 2a). The constructed neighbor-network based on the SNP data was also consistent with the genetic pattern evident from ADMIXTURE analyses and the PCA biplot. All the individuals of C. micrantha formed a single group that was positioned close to the group formed by individuals of C. “ptilosperma” (Fig. 2b). All C. “ptilosperma” individuals clustered at the intersection of the splits between C. micrantha and C. flavida (Fig. 2b).

Genetic analyses of C. micrantha, C. “ptilosperma” and C. flavida. (a) Principal component analysis (PCA) plot of the first two components. (b) Phylogenetic network showing genome reticulation. Branch lengths are proportional to absolute distances calculated from the binary matrix
Genetic diversity and differentiation statistics
The genetic diversity of C. flavida was estimated to be higher than that of either C. “ptilosperma” or C. micrantha (Table S1). All the C. flavida populations harbored extremely high within-population genetic variation, as reflected in all the measures of genetic variation estimated here; in stark contrast, genetic diversity was extremely low for all populations of C. micrantha (Table S1). The estimated FST values indicated a slightly low differentiation between C. “ptilosperma” and C. flavida (FST = 0.114). The genetic differentiation was greater between C. “ptilosperma” and C. micrantha (FST = 0.268) than between C. micrantha and C. flavida (FST = 0. 225) (Fig. 3a).

Genetic differentiation of C. micrantha, C. “ptilosperma” and C. flavida and the population history scenario examined in DIYABC. (a) FST within each taxa and between pairs of taxa based on the obtained genome-wide SNP data. (b) Scenario 1 was selected as being the most likely describe the origin of admixed lineages. Generations are shown on the y-axis (t0 to t4) and admixture proportions from each parent are shown on the scenario
ABC-based inferences of population history
In total, 161 individuals from 17 populations were examined. DIYABC analysis is based on a total of 415 SNPs after exclusion of monomorphic loci. The posterior probabilities for scenario 1 was 0.6317 (95% confidence interval (CI) 0.6097–0.6536), much higher than for the other nine scenarios (Fig. S2). For scenario 1, the median values of the effective population sizes of N1 (C. micrantha), N2 (C. “ptilosperma”), N3 (NG-LR and NG-GLA populations of C. flavida), N4 (C. flavida) and Na were 7.73 × 103, 3.30 × 103, 1.06 × 103, 1.25 × 104, 8.20 × 103, respectively (Fig. S3). The median values of the divergence time, t1, and the time of hybridization event, t2, were 97.9 (95% CI: 16.5–275) and 211 (95% CI: 42.8–1340) generations ago, respectively (Fig. S3). Principal component analyses showed that the summary statistics of observed datasets was similar to simulated datasets (Fig. S4), suggesting that scenario 1 was generally fitted to the observed data.
Results of approximate bayesian computation (ABC) approach showed that the most supported scenario (Scenario 1) was the one considering C. “ptilosperma” arose hybridization events between C. micrantha and C. flavida, and populations of NG-LR and NG-GLA was originated from introgression of C. micrantha genes into C. flavida (Fig. 3b).
Variation in pressure-volume (P-V) traits
Of all the populations of yellow camellias studied here, significant differences were observed in the leaf P-V parameters of C. “ptilosperma” vis-à-vis its putative parental species (Fig. 4a). The value of Ψ at turgor loss point (ΨTLP) for the C. “ptilosperma” was significantly more negative than that of either XH-NP (p = 0.01810) and XH-FL (p = 0.00002) populations of C. micrantha and likewise for the NG-LT (p = 0.00020) and NG-LR (p = 0.00041) populations of C. flavida. No significant differences were observed between the ΨTLP of C. micrantha and C. flavida (Fig. 4a).

The value of Ψ at turgor loss point (ΨTLP) and diameter of the vessels in the branches. (a) Comparison of the significantly negative value of ΨTLP of C. “ptilosperma” with the ΨTLP values of C. micrantha and C. flavida. * p < 0.05. (b) The diameter of the vessels in the branches of C. “ptilosperma” was significantly different from those of C. micrantha and C. flavida. * p < 0.05
Capacitance and wood properties
The water content of different Camellia taxa and tissues is presented in Table S2 and the corresponding water release curves of the branches and roots of the yellow Camellia species are depicted in Fig. S5. Branch or root capacitance did not differ significantly among the taxa studied (Table S2, Fig. S5). These results indicated that the yellow Camellia taxa we examined all have similar water storage capacity.
Nevertheless, the vessel diameters were narrowest in the branches of C. “ptilosperma”, and there were significant differences between the vessel diameters of C. “ptilosperma” in comparison with its putative parental species (Fig. 4b). Aside from that, however, there were negligible differences among these yellow Camellia taxa in their wood characteristics, including fibers/tracheids (F), and ray and axial parenchyma (RP and AP) (Table S3).
Discussion
Hybridization occurs between C. micrantha and C. flavida, resulting in C. “ptilosperma”
This study’s results supported naturally occurring hybridization and introgression between C. micrantha and C. flavida in narrow sympatric zone of southwest Guangxi (China), despite their strong morphological and ecological differentiation. The evidence for this came from a suite of complementary results for gene clustering, PCA, and the phylogenetic network analysis based on SNP data (Figs. 1 and 2).
In the ADMIXTURE analyses, population MZ-SJ of C. “ptilosperma” and the populations NG-LR and NG-GLA of C. flavida present a different degree of admixed ancestry and appear to constitute an independent cluster when K = 4 (Fig. 1j). However, a recent phylogenomics study revealed a conflicting relationship for C. “ptilosperma”, with it found more closely related to C. micrantha in the chloroplast phylogeny, whereas it was genetically closer to C. flavida in the phylogenetic trees constructed with nuclear DNA [45]. In contrast to C. “ptilosperma”, individuals from NG-LR and other populations of C. flavida show consistent relationships in all phylogenies [45]. Further, earlier it was found that only one chloroplast haplotype was observed in the C. “ptilosperma” population and it is genetically distinct from all haplotypes of C. flavida [55]. Altogether, this indicates that population MZ-SJ of C. “ptilosperma” and the populations NG-LR and NG-GLA of C. flavida have a distinct evolutionary origin. Approximate Bayesian computation result support scenario 1 as being the most likely one to have occurred, which suggests that hybridization events between C. micrantha and C. flavida gave rise to C. “ptilosperma”, and backcrossing led to the C. flavida populations NG-LR and NG-GLA being composed entirely of advanced of introgression individuals (Fig. 3b). When using K = 2, and thus intermediate to the parents, the ADMIXTURE analyses of both data sets demonstrated that C. “ptilosperma” showed greater similarity to C. micrantha (Fig. 1j). This result agrees with the expectation that the hybrid genome is unlikely to inherit an equal proportion from each parent as backcrossing is expected to occur [4]. Additionally, the different proportion of genetic material for the parental species to the genome of the hybrid could also be explained by the selection against minor parent ancestry [56].
Overall, population MZ-SJ (C. “ptilosperma”) is likely of hybrid origin and subsequently backcrossing with its parents, suggesting it could be a hybrid swarm. Specifically, C. micrantha served as the maternal parent while C. flavida served as the paternal parent during the initial hybridization event.
Novel hydraulic traits in C. “ptilosperma” support its distinct niche divergence
The ecophysiological experiments revealed that the focal taxa exhibited inherent differences in their hydraulic traits related to water availability. In particular, we find that the water potential at turgor loss point (ΨTLP) as well as vessel diameter in branches of C. “ptilosperma” differed significantly from its parental species.
Specifically, the ΨTLP values of C. “ptilosperma” were significantly lower than those of its parents (Fig. 4a). The maintenance of cell turgor in leaves is crucial for plants to avoid injury and/or for maintaining cellular functions during drought conditions [57, 58]. A more negative value of ΨTLP indicates a greater ability to withstand water deficits [59,60,61]. The significantly greater negative value of ΨTLP in C. “ptilosperma” suggests it has a survival advantage during the colonization of habitats where water availability is limited. We also observed an extremely narrow diameter of vessels in the branches of C. “ptilosperma” that was highly significantly different from those of its parental species (Fig. 4b). A similar pattern has been reported in other plant species growing in tropical karst forests [28]. This low vessel diameter indicates that C. “ptilosperma” compromises hydraulic efficiency for hydraulic safety, and this underlying mechanism could ensure survival in arid environments [62, 63].
Transgressive segregation is considered an important mechanism by which hybridization promotes ecological transition [4, 17, 18, 38, 64,65,66,67,68,69]. When hybrids exhibit novel or extreme characters vis-à-vis parental taxa, it is termed ‘transgressive segregation’, a phenomenon typically observed in early-generation hybrids [6, 54, 68]. For instance, in two alpine hybrid species their fixed novel drought-tolerance traits enable their persistence in the harsh alpine habitat [70, 71]. With the exception of genetic variation, phenotypic plasticity in hydraulic traits is another factor of adaptation [72,73,74]. Drought-tolerance traits in C. “ptilosperma” is also likely resulting from phenotypic plasticity. This emphasizes the need for conducting common garden experiments to confirm the origin of novel hydraulic traits in C. “ptilosperma”.
Our study demonstrated that compared with those of its parental species, C. “ptilosperma” had extreme ecophysiological traits related to drought tolerance. These findings suggest that C. “ptilosperma” has acquired novel traits lacking in either parent, and which are consistent with its adaptation to niches that are unfavorable to both its parental species.
Hybridization as a mechanism driving lineage diversification in yellow camellias
Hybridization is considered a producer of genetic variation that provides species at early stages of differentiation with prerequisite evolutionary novelties [12, 75, 76]. Such novel phenotypes may facilitate adaptive invasion of hybrid organisms into novel ecological niches, which enables ecological differentiation between hybrid progenies and parental species [4]. Additionally, divergent phenology can also decrease gene flow between hybrids and parents, thereby favoring hybrid establishment and subsequent independent evolution [12, 17, 65].
The hybrid derivative C. “ptilosperma” appears to conform to this expectation. Indeed, our study provides substantial, compelling evidence regarding the hybrid origin of C. “ptilosperma”. Ecophysiological results also confirm that extreme characters (significantly negative value of ΨTLP and smaller vessel diameter) have become well fixed in C. “ptilosperma”, conferring to it a greater ability to withstand drought, thus allowing it to grow on karst hillslopes where they must rely on shallow soil water. Moreover, C. “ptilosperma” differs in a key phenological trait from its ancestral parents. Best known for its long flowering period (from May to December, with the peak of flowering in July) [49], C. “ptilosperma” had been considered as the candidate for generating new cultivated varieties in yellow-flower Camellia breeding [77, 78]. Interestingly, C. micrantha plants flower from November to December (peak flowering in November), and C. flavida flowers from September to November (peak flowering in September to October). This phenological difference is genetically based, as it persists for these plants when growing in a common garden of Golden Camellia Park in Nanning, Guangxi. Accordingly, the ecological novelty and distinct phenology attribute of C. “ptilosperma” most likely arose through the combination of genetic material from those two parents.
Notably, the specific traits found in C. “ptilosperma” could serve as an effective barrier to genetic exchange with its parents, contributing to its persistence, and thus promoting conditions for the establishment a new adaptive lineage. Likewise, limestone karst areas provide an essential habitat suite for the generation of hybrid plants, given that novel ecological opportunities are crucial for the origin of hybrid species [14, 17]. Karst landforms are generally covered by discontinuous and thin soils, with a high infiltration and underground drainage system, resulting in the creation of a variety of microhabitats [51]. Soil nutrients and moisture were heterogeneity along the topography of karst ecosystems [23, 46, 79, 80]. The complex karst terrain provides various vacant ecological niches for hybrids to avoid competition and/or gene flow from their parental populations.
Conclusion
We found evidence for natural hybridization between C. micrantha and C. flavida that gave rise to hybrid population, C. “ptilosperma”. The novel hydraulic traits fixed in C. “ptilosperma” explain its distinct ecological niche, which lies outside its parental ranges. We highlight the role of hybridization in facilitating the evolution of hybrid population with unique phenological and ecological. This study reveals a case of natural hybridization that facilitated lineage diversification in yellow camellias.
Methods
Population sampling
A total of 10 populations of C. flavida (n = 91 individuals) and 6 populations of C. micrantha (n = 53 individuals) were sampled from their natural collection sites (hereafter referred to as populations) in southwestern Guangxi, China (Fig. 1a). The samples collected spanned the entire distribution range of either species. Unfortunately, wild populations of MZ-SJ (C. “ptilosperma”) have been severely destroyed, the samples for it used in this study could only be collected from just 17 individuals. All the studied taxa were diploid (2n = 30) [81].
DNA extraction, library preparation and sequencing
Fresh leaf tissues were collected from all the sampled plants and preserved on dry ice for sequencing. Total genomic DNA was extracted from each tissue sample by using a modified cetyltrimethylammonium bromide (CTAB) protocol [82] and then double digested with the restriction enzymes EcoRI and NIaIII. Paired-end 150 bp reads were sequenced using a NovaSeq 6000 (Illumina) platform. The raw reads obtained from each sample were deposited at the NCBI Sequence Read Archive (SRA) database (accession number: PRJNA770534).
Bioinformatic data processing
The sequences were assembled into de novo loci using the STACKS v2.54 pipeline [83] for obtaining the SNP data. The process_radtag module of STACKS was used for quality-filtered and demultiplexing the raw fastq reads. In order to remove the tags with intact restriction enzyme cut sites and ambiguous barcode near the 3′ ends, the sequences were trimmed to a length of 135 bp using the SEQTK tool [84]. The clean reads thus obtained were further assembled to stacks (ustacks), which were used to build a catalog (cstacks) for variant calling. In these two steps, two main parameters (M parameter of ustacks, and n parameter of cstacks) were identical to those suggested previously for use in phylogenomics of yellow camellias [45], which were determined based on the protocol described by Rochette and Catchen [85]. All the samples in the population map were matched against the catalog using the sstacks. Pair-end reads were associated with each single-end locus using tsv2bam and assembled into contigs using gstacks. The output SNP data files were exported using the populations in STACKS. The loci that were sequenced from at least 80% of the individuals in each population (-r 0.8) in at least 13 populations (-p 13) were retained.
The original variants were filtered for generating a final SNP dataset using the VCFtools v0.1.16 software [86]. The following criteria were used for generating the SNP dataset: (1) SNPs with minimum minor allele frequency of 0.05, SNPs with less than 20% missing data, and only biallelic sites were retained; (2) markers significantly deviated from Hardy-Weinberg equilibrium (p < 0.05) were exclude.
Population structure and admixture
A population genomic approach was employed for analyzing the population genetic structure in this study, and the hybrids were identified using the ADMIXTURE program [87]. The analysis was repeated 100 times, from K = 2 to K = 10. We ran a fivefold cross-validation (CV) to assessed the best fit number of clusters (based on the K value). Apart from clustering analyses with the ADMIXTURE program, principal component analysis (PCA) was performed in this study using the EIGENSOFT package of the smartpca program [88], and the results were visualized using R v3.6.3 (https://www.r-project.org/). The pattern of genetic clustering was subsequently visualized by creating population splits based on the uncorrected P-distance between individuals using the Neighbor-Net algorithm of SplitTree4 program [89].
Genetic statistics
Genetic statistics, including the average values of major allele frequency (P), observed heterozygosity (HO), expected heterozygosity (HE), and nucleotide diversity (π) across loci, were estimated using STACKS v2.54 [83]. Population differentiation within and between species were measured by the pairwise genetic differentiation parameter (FST), and the significance of the observed FST was determined using 10,000 permutations in the Arlequin program [90].
Testing hybrid origins with ABC
Based on the ADMIXTURE, PCA and Neighbor-net tree results, four genetic group were defined. These consisted of C. micrantha, a population referred to as C. “ptilosperma” (i.e., population MZ-SJ), two intraspecific lineages of C. flavida that included the populations NG-LR and NG-GLA, and all remaining populations. We used approximate bayesian computation (ABC) to compare different evolutionary scenarios for the origin of population MZ-SJ and populations NG-LR and NG-GLA of C. flavida using the software package DIYABC v.2.1.0 [91]. A total of ten scenarios were tested (Fig. S2). Under scenarios 1, 2, 3 and 4, the lineages diverge from C. micrantha/C. flavida and hybridize with the second species. Under scenarios 5 and 6, the MZ-SJ population/C. flavida populations NG-LR and NG-GLA originate from crosses between C. micrantha and C. flavida then respectively follow introgression from C. micrantha to C. flavida. Under scenarios 7 and 8, the lineages arise from a single origin based on a hybridization event between C. micrantha and C. flavida (the MZ-SJ population and C. flavida’s NG-LR and NG-GLA populations diverge due to cladogenesis from this hybrid lineage), whereas they diverge from C. flavida under scenarios 9 and 10, respectively.
The variant call format (VCF) file of the unlinked SNPs was converted to DIYABC format using the Python script vcf2DIYABC.py available from https://github.com/loire/vcf2DIYABC.py. To meet the requirements of DIYABC, SNPs were removed if they include monomorphic loci. For each prior, we set the interval for population sizes and divergence time to 10–105, and that for admixture rates to 0.001–0.999, because we lacked sufficient knowledge about the population sizes, divergence times, and admixture rate of Camellia species. A total of 25 summary statistics including genetic diversity, pairwise sample FST, and Nei’s distance as well as various admixture summary statistics were used to compare the observed versus simulated data [92]. For each scenario 106 simulations were performed, from which we estimated parameter posterior distributions by taking the 1% of simulated datasets closest to the observed dataset for use in local linear regression. To compare the posterior probability of the 10 scenarios, the 10 000 (1%) simulated datasets closest to the observed dataset were selected for use in logistic regression and 500 for use with the direct approach.
After choosing the best model, the posterior distribution of each parameter was also estimated by taking 1 000 (1%) simulated datasets closet to the observed dataset for the local linear regression and applying a logit transformation to a given parameter’s values. The goodness-of-fit of the tested scenarios was assessed implementing the model-checking function of DIYABC. The PCA was carried out to visually assess the position of the observed dataset vis-à-vis the simulated datasets.
Ecophysiological traits
The study site was located at Nonggang National Natural Reserve, **xiang Munipality and Daqing Mountain, Longzhou county, Guangxi, Southern China (21.82° – 22.53° N, 106.74° – 107.23° E). The study region is profoundly influenced by the subtropical monsoon. The mean annual temperature is 22 °C; precipitation ranges from 1150 to l550 mm per year, interrupted by a dry season from October to April [26]. The selection of samples used for hydraulic measurements was based on the results from the ADMIXTURE program (using K = 2), and consisted of individuals from populations of C. flavida (NG-LT and NG-LR), C. micrantha (XH-BS and BY-NP), and C. “ptilosperma” (MZ-SJ). Ecophysiological trait data were obtained from three sets of measurements. The first being leaf pressure–volume (P–V) curves obtained for each taxon to determine the relation of its leaf-level water potential at the turgor loss point to physiological drought tolerance. The second was hydraulic capacitance, assumed here to be associated with water storage strategy. The third set of measurements comprised wood anatomy traits, which are often tightly linked to the water availability of plants. We measured all the hydraulic traits of plants during the dry season (in October 2021). All measurements were performed in the laboratory of Experimental Center of Tropical Forests, Chinese Academy of Forestry (**xiang Munipality, Guangxi, China). Materials were moved into the laboratory within an hour of sampling.
Leaf pressure- volume (P-V) curves
The leaf P-V curves were generated using the bench drying method [93]. For each taxon, five short leaf-bearing, sun-exposed branches were selected from five mature individuals in the early morning (one per individual plant). The collected branches were sealed in black plastic bags after cut off under water in a bucket, and immediately transported to the laboratory. A segment of approximately 10 cm was removed from the cut end under water. The PMS-1505D pressure chamber (Corvallis, OR, USA) was used for determining the water potential after 2 h of rehydration. The weight and water potential of the leaves were periodically estimated during dehydration. The final dry weight of the samples was determined by oven drying at 60 °C for 48 h. The weight of the leaves (g) was measured using a balance, and the leaf area (m2) was measured using a scanner.
Measurement of capacitance
The capacitance was measured according to the methods described by Jupa, Plavcová, Gloser and Jansen [94]. Five old branches or roots, approximately 1 cm in diameter and 15 cm in length, were collected from five mature and healthy individuals per taxon. Freshly excised segments of branches and roots were wrapped in a plastic bag and immediately transported to the laboratory for further analyses. After removing the bark, the cambium and pith were separated from the segments, and the remaining tissues were cut into 2-cm-long segments and vacuum- infiltrated with distilled water for 12 h. After rehydration, the remaining segments were shortened to a 5-mm length to quantify water potential with a WP4C water potential meter (Meter, Hopkinsville, USA); this apparatus was calibrated prior to measurements on a daily basis. The weight of the segments and their water potential were periodically measured during dehydration stage until the water potential fell below − 8 MPa. Next, the volume of each sample was determined using the water displacement method, after which all samples were oven-dried at 70 °C for at least 48 h for their dry mass determination. Relative water content (RWC) was calculated using this equation:
where Wf and Wd denote the weight of a given sample before and after its dehydration, respectively, and Ws is the weight of the sample when fully saturated. Cumulative water release (CWR, kg m− 3) was calculated as follows:
where \({\rho }\) is the density of the wood (g/cm− 3), this expressed as the ratio of dry mass of a sample to its volume. To describe the relationship between the CWR and water potential (Ψ) of each Camellia species, the data were fitted to the hyperbolic function:
where a is the best-fitting parameter denoting the value of CWR in the asymptotic region of the curve, and b is the best-fitting parameter for the values of Ψ. The shape of the curve was divided into two phases according to the b parameter value: phase I (from Ψ = 0 to b MPa) and phase II (from Ψ = b to − 8 MPa). Phase I was characterized by the initial rapid release of water, concurrent with a decline in Ψ, and phase II corresponded to the period of gradual water release. Finally, the capacitance in the two distinct phases was calculated as the ratio of ∆CWR to ∆Ψ:
Wood anatomy
After measuring the hydraulic capacitance, the remaining fresh branches and roots were used to analyze the anatomical characteristics of each sample’s woody tissue. To do this, transverse sections (approximately 25 μm) were prepared using a sliding microtome (SM2010R, Leica, Wetzlar, Germany). under a light microscope (Nikon Eclipse 50i, Nikon, Tokyo, Japan) and the obtained images then analyzed in ImageJ software [95]. vessel diameter (D) and relative proportion of various cell types in the wood, namely, vessels (V), fibers (including tracheids) (F), and ray and axial parenchyma (RP and AP), were determined from more than 10 fields per transverse section. In Fig. 5 are representative images of wood anatomy obtained by light microscopy.

Representative light microscopy images of transverse sections of wood anatomy of the yellow camellias studied herein. Axial parenchyma (AP), ray parenchyma (RP), vessel (V), fiber (F)
Data Availability
statement.
The datasets generated or analysed during the current study are available in the NCBI Sequence Read Archive and can be found under BioProject PRJNA770534 with accession nos. SAMN30101064–SAMN30101208, SAMN22253095–SAMN22253102, SAMN22253085–SAMN22253086, SAMN22253119–SAMN22253120, SAMN22253123–SAMN22253126 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA770534).
Abbreviations
- ABC:
-
approximate bayesian computation
- AP:
-
axial parenchyma
- CTAB:
-
cetyltrimethylammonium bromide
- CV:
-
cross-validation
- CI:
-
confidence interval
- D:
-
diameter
- ddRAD:
-
double digest restriction-site associated DNA
- F ST :
-
genetic differentiation index
- F:
-
fibers
- H O :
-
observed heterozygosity
- H E :
-
expected heterozygosity
- π:
-
nucleotide diversity
- P:
-
major allele frequency
- PCA:
-
principal component analysis
- P–V:
-
pressure–volume
- RP:
-
ray parenchyma
- SNP:
-
single nucleotide polymorphism
- SD:
-
standard deviation
- SRA:
-
Sequence Read Archive
- VCF:
-
variant call format
- V:
-
vessels
- WGD:
-
whole genome duplication
- RWC:
-
relative water content
- Ψ:
-
water potential
- ΨTLP :
-
the water potential at turgor loss point
References
Soltis PS, Soltis DE. The role of hybridization in plant speciation. Annu Rev Plant Biol. 2009;60:561–88. https://doi.org/10.1146/annurev.arplant.043008.092039
Pennisi E. Shaking up the tree of life. Science. 2016. https://doi.org/10.1126/science.354.6314.817. https://www.science.org/doi/
Abbott RJ, Barton NH, Good JM. Genomics of hybridization and its evolutionary consequences. Mol Ecol. 2016. https://doi.org/10.1111/mec.13685
Mallet J, Hybrid speciation. Nature. 2007;446(7133):279–83. https://doi.org/10.1038/nature05706
Rieseberg LH, Willis JH. Plant speciation. Science. 2007;317(5840):910–4.
Seehausen O. Hybridization and adaptive radiation. Trends Ecol Evol. 2004;19(4):198–207. https://doi.org/10.1016/j.tree.2004.01.003Get
Hamilton JA, Miller JM. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv Biol. 2016;30(1):33–41. https://doi.org/10.1111/cobi.12574
Ma Y, Mao X, Wang J, Zhang L, Jiang Y, Geng Y, Ma T, Cai L, Huang S, Hollingsworth P. Pervasive hybridization during evolutionary radiation of Rhododendron subgenus Hymenanthes in mountains of southwest China. Natl Sci Rev. 2022;9(12):nwac276. https://doi.org/10.1093/nsr/nwac276
Jorgensen TH, Olesen JM. Adaptive radiation of island plants: evidence from Aeonium (Crassulaceae) of the Canary Islands. Perspect Plant Ecol. 2001;4(1):29–42. https://doi.org/10.1078/1433-8319-00013
Svardal H, Quah FX, Malinsky M, Ngatunga BP, Miska EA, Salzburger W, Genner MJ, Turner GF, Durbin R. Ancestral hybridization facilitated species diversification in the Lake Malawi cichlid fish adaptive radiation. Mol Biol Evol. 2020;37(4):1100–13. https://doi.org/10.1093/molbev/msz294
Givnish TJ. Adaptive radiation versus ‘radiation’and ‘explosive diversification’: why conceptual distinctions are fundamental to understanding evolution. New Phytol. 2015;207(2):297–303. https://doi.org/10.1111/nph.13482
Nolte AW, Tautz D. Understanding the onset of hybrid speciation. Trends Genet. 2010;26(2):54–8. https://doi.org/10.1016/j.tig.2009.12.001
Burke JM, Arnold ML. Genetics and the fitness of hybrids. Annu Rev Genet. 2001;35(1):31–52.
Buerkle CA, Morris RJ, Asmussen MA, Rieseberg LH. The likelihood of homoploid hybrid speciation. Heredity. 2000;84(4):441–51. https://doi.org/10.1046/j.1365-2540.2000.00680.x
Suarez-Gonzalez A, Lexer C, Cronk QC. Adaptive introgression: a plant perspective. Biol Lett. 2018;14(3):20170688. https://doi.org/10.1098/rsbl.2017.0688
Ma Y, Wang J, Hu Q, Li J, Sun Y, Zhang L, Abbott RJ, Liu J, Mao K. Ancient introgression drives adaptation to cooler and drier mountain habitats in a cypress species complex. Commun Biol. 2019;2(1):1–12. https://doi.org/10.1038/s42003-019-0445-z
Gross BL, Rieseberg L. The ecological genetics of homoploid hybrid speciation. J Hered. 2005;96(3):241–52. https://doi.org/10.1093/jhered/esi026
Lexer C, Welch ME, Raymond O, Rieseberg LH. The origin of ecological divergence in Helianthus paradoxus (Asteraceae): selection on transgressive characters in a novel hybrid habitat. Evolution. 2003;57(9):1989–2000. https://doi.org/10.1111/j.0014-3820.2003.tb00379.x
Lowry DB, Modliszewski JL, Wright KM, Wu CA, Willis JH. The strength and genetic basis of reproductive isolating barriers in flowering plants. Phil Trans R Soc B. 2008;363(1506):3009–21. https://doi.org/10.1098/rstb.2008.0064
Hall MC, Willis JH. Divergent selection on flowering time contributes to local adaptation in Mimulus guttatus populations. Evolution. 2007;60(12):2466–77. https://doi.org/10.1111/j.0014-3820.2006.tb01882.x
Marques DA, Meier JI, Seehausen O. A combinatorial view on speciation and adaptive radiation. Trends Ecol Evol. 2019;34(6):531–44. https://doi.org/10.1016/j.tree.2019.02.008
Clements R, Sodhi NS, Schilthuizen M, Ng PK. Limestone karsts of Southeast Asia: imperiled arks of biodiversity. Bioscience. 2006;56(9):733–42. https://doi.org/10.1641/0006-3568(2006)56[733:LKOSAI]2.0.CO;2
Geekiyanage N, Goodale UM, Cao K, Kitajima K. Plant ecology of tropical and subtropical karst ecosystems. Biotropica. 2019;51(5):626–40. https://doi.org/10.1111/btp.12696
Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403(6772):853–8. https://doi.org/10.1038/35002501
Guo YL, Wang B, Mallik AU, Huang F, **ang WS, Ding T, Wen SJ, Lu SH, Li DX, He YL. Topographic species–habitat associations of tree species in a heterogeneous tropical karst seasonal rain forest, China. J Plant Ecol. 2017;10(3):450–60. https://doi.org/10.1093/jpe/rtw057
Guo YL, Wang B, Li DX, Mallik AU, **ang WS, Ding T, Wen SJ, Lu SH, Huang FZ, He YL. Effects of topography and spatial processes on structuring tree species composition in a diverse heterogeneous tropical karst seasonal rainforest. Flora. 2017;231:21–8. https://doi.org/10.1016/j.flora.2017.04.002
Zhang QW, Zhu SD, Jansen S, Cao KF. Topography strongly affects drought stress and xylem embolism resistance in woody plants from a karst forest in Southwest China. Funct Ecol. 2021;35(3):566–77. https://doi.org/10.1111/1365-2435.13731
Zhu SD, Chen YJ, Fu PL, Cao KF. Different hydraulic traits of woody plants from tropical forests with contrasting soil water availability. Tree Physiol. 2017;37(11):1469–77. https://doi.org/10.1093/treephys/tpx094
Chung KF, Leong WC, Rubite RR, Repin R, Kiew R, Liu Y, Peng CI. Phylogenetic analyses of Begonia sect. Coelocentrum and allied limestone species of China shed light on the evolution of sino-vietnamese karst flora. Bot Stud. 2014;55(1):1–15. https://doi.org/10.1186/1999-3110-55-1
Kang M, Tao J, Wang J, Ren C, Qi Q, **ang QY, Huang HW. Adaptive and nonadaptive genome size evolution in Karst endemic flora of China. New Phytol. 2014;202(4):1371–81. https://doi.org/10.1111/nph.12726
Wang J, Ai B, Kong HH, Kang M. Speciation history of a species complex of Primulina eburnea (Gesneriaceae) from limestone karsts of southern China, a biodiversity hot spot. Evol Appl. 2017;10(9):919–34. https://doi.org/10.1111/eva.12495
Wang J, Feng C, Jiao TL, Von Wettberg EB, Kang M. Genomic signature of adaptive divergence despite strong nonadaptive forces on edaphic islands: a case study of Primulina juliae. Genome Biol Evol. 2017;9(12):3495–508. https://doi.org/10.1093/gbe/evx263
Gao Y, Ai B, Kong HH, Kang M, Huang HW. Geographical pattern of isolation and diversification in karst habitat islands: a case study in the Primulina eburnea complex. J Biogeogr. 2015;42(11):2131–44. https://doi.org/10.1111/jbi.12576
Feng C, Wang J, Wu LQ, Kong HH, Yang LH, Feng C, Wang K, Rausher M, Kang M. The genome of a cave plant, Primulina huaijiensis, provides insights into adaptation to limestone karst habitats. New Phytol. 2020;227(4):1249–63. https://doi.org/10.1111/nph.16588
Tian DK, **ao Y, Tong Y, Fu NF, Liu QQ, Li C. Diversity and conservation of chinese wild begonias. Plant Divers. 2018;40(3):75–90. https://doi.org/10.1016/j.pld.2018.06.002
Hughes M, Rubite RR, Blanc P, Chung KF, Peng CI. The Miocene to Pleistocene colonization of the philippine archipelago by Begonia sect. Baryandra (Begoniaceae). Am J Bot. 2015;102(5):695–706. https://doi.org/10.3732/ajb.1400428
Tian DK, Li C, **ao Y, Fu NF, Tong Y, Wu RJ. Occurrence and characteristics of natural hybridization in Begonia in China. Biodivers. 2017;25(6):654–74. https://doi.org/10.17520/biods.2017050
Rieseberg LH, Kim SC, Randell RA, Whitney KD, Gross BL, Lexer C, Clay K. Hybridization and the colonization of novel habitats by annual sunflowers. Genetica. 2007;129(2):149–65. https://doi.org/10.1007/s10709-006-9011-y
Chang HT, Ren SX. Theaceae. In: Chang HT, editor. Flora Reipublicae Popularis Sinicae. Bei**g: Science Press; 1998. pp. 101–10.
Chang HT, Yang CH, Zhang TZ. A new species of yellow Camellia from Guizhou. Guihaia. 1997;17(4):289–90.
Huang LD, Liang SY, Ye CX. A new species of Camellia from Guangxi. Guangdong Landsc Archit. 2014;36(1):69–70.
Hu RC, Wei SJ, Liufu YQ, Nong YK, Fang W. Camellia debaoensis (Theaceae), a new species of yellow camellia from limestone karsts in southwestern China. PhytoKeys. 2019;135:49. https://doi.org/10.3897/phytokeys.135.38756
Liu ZW, Fang W, Liu ED, Zhao M, He YF, Yang SX. Camellia mingii, a new species of yellow camellias from Southeast Yunnan, China. Phytotaxa. 2019;393(1):47–56. https://doi.org/10.11646/phytotaxa.393.1.4
Liu ZW, Chai SF, Wu FY, Ye PM, Jiang CJ, Zhang ZR, Yu XQ, Ma JL, Yang SX. Camellia rostrata, a new species of yellow camellias from Southwest China. Phytotaxa. 2020;459(1):061–8. https://doi.org/10.11646/phytotaxa.459.1.6
Wei S, Liufu Y, Zheng H, Chen H, Lai Y, Liu Y, Ye Q, Tang S. Using phylogenomics to untangle the taxonomic incongruence of yellow-flowered Camellia species (Theaceae) in China. J Syst Evol. 2022. https://doi.org/10.1111/jse.12915
Hao Z, Kuang Y, Kang M. Untangling the influence of phylogeny, soil and climate on leaf element concentrations in a biodiversity hotspot. Funct Ecol. 2015;29(2):165–76. https://doi.org/10.1111/1365-2435.12344
Fu PL, Jiang YJ, Wang AY, Brodribb TJ, Zhang JL, Zhu SD, Cao KF. Stem hydraulic traits and leaf water-stress tolerance are co-ordinated with the leaf phenology of angiosperm trees in an asian tropical dry karst forest. Ann Bot. 2012;110(1):189–99. https://doi.org/10.1093/aob/mcs092
Fu PL, Liu WJ, Fan ZX, Cao KF. Is fog an important water source for woody plants in an asian tropical karst forest during the dry season? Ecohydrology. 2016;9(6):964–72. https://doi.org/10.1002/eco.1694
Liang SY. Two new species of Camellia from Guangxi, China. Bull Bot Res. 1984;4:183–8.
Ming TL, Bartholomew B. Theaceae. In: Wu ZY, Raven PH, editors. Flora of China. Bei**g: Science Press; 2007. pp. 368–72.
Hartmann A, Goldscheider N, Wagener T, Lange J, Weiler M. Karst water resources in a changing world: review of hydrological modeling approaches. Rev Geophys. 2014;52(3):218–42. https://doi.org/10.1002/2013RG000443
Willimas PW. The role of the epikarst in karst and cave hydrogeology: a review. Int J Speleol. 2008;37:1–10. https://doi.org/10.5038/1827-806X.37.1.1
Rieseberg LH. Hybrid origins of plant species. Annu Rev Ecol Syst. 1997;28(1):359–89. http://www.jstor.org/stable/2952498
Rieseberg LH, Archer MA, Wayne RK. Transgressive segregation, adaptation and speciation. Heredity. 1999;83(4):363–72. https://doi.org/10.1038/sj.hdy.6886170
Wei SJ, Lu YB, Ye QQ, Tang SQ. Population genetic structure and phylogeography of Camellia flavida (Theaceae) based on chloroplast and nuclear DNA sequences. Fron Plant Sci. 2017;8:718. https://doi.org/10.3389/fpls.2017.00718
Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science. 2018;360(6389):656–60.
Scoffoni C, Vuong C, Diep S, Cochard H, Sack L. Leaf shrinkage with dehydration: coordination with hydraulic vulnerability and drought tolerance. Plant Physiol. 2014;164(4):1772–88. https://doi.org/10.1104/pp.113.221424
Kozlowski TT, Pallardy S. Acclimation and adaptive responses of woody plants to environmental stresses. Bot Rev. 2002;68(2):270–334. https://doi.org/10.1663/0006-8101(2002. )068[0270:AAAROW]2.0.CO;2.
Zhu SD, Chen YJ, Ye Q, He PC, Liu H, Li RH, Fu PL, Jiang GF, Cao KF. Leaf turgor loss point is correlated with drought tolerance and leaf carbon economics traits. Tree Physiol. 2018;38(5):658–63. https://doi.org/10.1093/treephys/tpy013
Blackman CJ. Leaf turgor loss as a predictor of plant drought response strategies. Tree Physiol. 2018;38(5):655–7. https://doi.org/10.1093/treephys/tpy047
Baltzer JL, Davies SJ, Bunyavejchewin S, Noor N. The role of desiccation tolerance in determining tree species distributions along the Malay–Thai Peninsula. Funct Ecol. 2008;22(2):221–31. https://doi.org/10.1111/j.1365-2435.2007.01374.x
Sperry JS, Meinzer FC, McCulloh KA. Safety and efficiency conflicts in hydraulic architecture: scaling from tissues to trees. Plant Cell Environ. 2008;31(5):632–45. https://doi.org/10.1111/j.1365-3040.2007.01765.x
Pfautsch S, Harbusch M, Wesolowski A, Smith R, Macfarlane C, Tjoelker MG, Reich PB, Adams MA. Climate determines vascular traits in the ecologically diverse genus Eucalyptus. Ecol Lett. 2016;19(3):240–8. https://doi.org/10.1111/ele.12559
Kagawa K, Takimoto G. Hybridization can promote adaptive radiation by means of transgressive segregation. Ecol Lett. 2018;21(2):264–74. https://doi.org/10.1111/ele.12891
Abbott RJ, Hegarty MJ, Hiscock SJ, Brennan AC. Homoploid hybrid speciation in action. Taxon. 2010;59(5):1375–86. https://doi.org/10.1002/tax.595005
Dittrich-Reed DR, Fitzpatrick BM. Transgressive hybrids as hopeful monsters. Evol Bioll. 2013;40:310–5.
Bell MA, Travis MP. Hybridization, transgressive segregation, genetic covariation, and adaptive radiation. Trends Ecol Evol. 2005;20(7):358–61. https://doi.org/10.1016/j.tree.2005.04.021
Rieseberg LH, Widmer A, Arntz AM, Burke B. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philosophical Trans Royal Soc Lond Ser B: Biol Sci. 2003;358(1434):1141–7.
Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C. Major ecological transitions in wild sunflowers facilitated by hybridization. Science. 2003;301(5637):1211–6. https://doi.org/10.1126/science.1086949
Wang J, Wang M, Zhang X, Sun S, Zhang A, Chen N, Zhao C. Enhanced cell dehydration tolerance and photosystem stability facilitate the occupation of cold alpine habitats by a homoploid hybrid species, Picea purpurea. AoB Plants. 2018;10(5):ply053. https://doi.org/10.1093/aobpla/ply053
Ma F, Zhao C, Milne R, Ji M, Chen L, Liu J. Enhanced drought-tolerance in the homoploid hybrid species Pinus densata: implication for its habitat divergence from two progenitors. New Phytol. 2010;185(1):204–16. https://doi.org/10.1111/j.1469-8137.2009.03037.x
Skelton RP, Anderegg LD, Papper P, Reich E, Dawson TE, Kling M, Thompson SE, Diaz J, Ackerly DD. No local adaptation in leaf or stem xylem vulnerability to embolism, but consistent vulnerability segmentation in a north american oak. New Phytol. 2019;223(3):1296–306.
Lamy JB, Delzon S, Bouche PS, Alia R, Vendramin GG, Cochard H, Plomion C. Limited genetic variability and phenotypic plasticity detected for cavitation resistance in a M editerranean pine. New Phytol. 2014;201(3):874–86.
Stojnić S, Suchocka M, Benito-Garzón M, Torres-Ruiz J, Cochard H, Bolte A, Cocozza C, Cvjetković B, De Luis M, Martinez-Vilalta J. Variation in xylem vulnerability to embolism in european beech from geographically marginal populations. Tree Physiol. 2018;38(2):173–85.
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R, et al. Hybridization and speciation. J Evol Biol. 2013;26(2):229–46. https://doi.org/10.1111/j.1420-9101.2012.02599.x
Soltis P. Hybridization, speciation and novelty. J Evol Biol. 2013;26(2):291–3.
You MX, Wang J, You MF, Huang LD. New hope for breeding new varieties with Camellia chuangtsoensis. Int Camellia J. 2013;45:97–100.
Cheng JS, Chen JY, Zhao SW. Interspecific cross breeding for new yellow Camellia. J Bei**g Forestry Univ. 1994;16:55–9.
Geekiyanage N, Goodale UM, Cao K, Kitajima KJB. Leaf trait variations associated with habitat affinity of tropical karst tree species. Ecol Evol. 2018;8(1):286–95. https://doi.org/10.1002/ece3.3611
Fu T, Chen H, Fu Z, Wang K. Surface soil water content and its controlling factors in a small karst catchment. Environ Earth Sci. 2016;75(21):1–11.
Zhang WJ, Ming TL. Karyotypical study of sect. Archecamellia of genus Camellia. Plant Divers. 1995;17(01):1.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–9.
Rochette NC, Rivera-Colón AG, Catchen JM. Stacks 2: Analytical methods for paired‐end sequencing improve RADseq‐based population genomics. Mol Ecol. 2019;28(21):4737–54. https://doi.org/10.1111/mec.15253
Li H. Toolkit for processing sequences in FASTA/Q formats. https://github.com/lh3/seqtk. Accessed 18 Jun 2018.
Rochette NC, Catchen JM. Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc. 2017;12(12):2640–59. https://doi.org/10.1038/nprot.2017.123
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. https://doi.org/10.1093/bioinformatics/btr330
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. http://www.genome.org/cgi/doi/10.1101/gr.094052.109
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. https://doi.org/10.1371/journal.pgen.0020190
Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67. https://doi.org/10.1093/molbev/msj030
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:117693430500100003. https://doi.org/10.1177/117693430500100003
Cornuet J-M, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin J-M, Estoup A. DIYABC v2. 0: a software to make approximate bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics. 2014;30(8):1187–9. https://doi.org/10.1093/bioinformatics/btt763
Choisy M, Franck P, Cornuet JM. Estimating admixture proportions with microsatellites: comparison of methods based on simulated data. Mol Ecol. 2004;13(4):955–68. https://doi.org/10.1111/j.1365-294X.2004.02107.x
Tyree M, Hammel H. The measurement of the turgor pressure and the water relations of plants by the pressure-bomb technique. J Exp Bot. 1972;23(1):267–82. https://doi.org/10.1093/jxb/23.1.267
Jupa R, Plavcová L, Gloser V, Jansen S. Linking xylem water storage with anatomical parameters in five temperate tree species. Tree Physiol. 2016;36(6):756–69. https://doi.org/10.1093/treephys/tpw020
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. https://doi.org/10.1038/nmeth.2019
Acknowledgements
We thank Hailing Chen, Yanchi Lai and Hewen Zheng for providing field assistance. We thank Nonggang National Nature Reserve for help in sample collection. We thank Experimental Center of Tropical Forests, Chinese Academy of Forestry for permission to conduct the ecophysiological experiments. We thank National Earth System Science Data Center, National Science & Technology Infrastructure of China for the data support.
Funding
This study was supported by the National Natural Science Foundation of China (grant number 31760088) and the Guangdong Provincial Special Research Grant for the Creation of National Parks (2021GJGY034).
Author information
Authors and Affiliations
Contributions
S.T. and W.L. designed the study. S.W. and Q.Z. performed experiments and analyzed the data. S.W., W.L. and S.T. wrote and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Materials collection, sequencing and all experiments were carried out in strict accordance with local laws and relevant laboratory regulations. S.T. undertook the formal identification of the plant material used in this study. Voucher specimens were deposited in the Herbarium of Guangxi Institute of Botany (IBK) under the voucher numbers IBK00439687 (population XH-BS of Camellia micrantha), IBK00439697 (population XH-NP of Camellia micrantha), IBK00425111 (population MZ-SJ of Camellia ptilosperma), IBK00425126 (population NG-LR of Camellia flavida) and IBK00441171 (population NG-LT of Camellia flavida).
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wei, S., Zhang, Q., Tang, S. et al. Genetic and ecophysiological evidence that hybridization facilitated lineage diversification in yellow Camellia (Theaceae) species: a case study of natural hybridization between C. micrantha and C. flavida. BMC Plant Biol 23, 154 (2023). https://doi.org/10.1186/s12870-023-04164-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04164-4