
Abstract
Background
Phosphorus (P) is an essential macronutrient for plant growth that participates in a series of biological processes. Thus, P deficiency limits crop growth and yield. Although Stylosanthes guianensis (stylo) is an important tropical legume that displays adaptation to low phosphate (Pi) availability, its adaptive mechanisms remain largely unknown.
Results
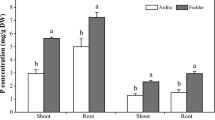
In this study, differences in low-P stress tolerance were investigated using two stylo cultivars (‘RY2’ and ‘RY5’) that were grown in hydroponics. Results showed that cultivar RY2 was better adapted to Pi starvation than RY5, as reflected by lower values of relative decrease rates of growth parameters than RY5 at low-P stress, especially for the reduction of shoot and root dry weight. Furthermore, RY2 exhibited higher P acquisition efficiency than RY5 under the same P treatment, although P utilization efficiency was similar between the two cultivars. In addition, better root growth performance and higher leaf and root APase activities were observed with RY2 compared to RY5. Subsequent RNA-seq analysis revealed 8,348 genes that were differentially expressed under P deficient and sufficient conditions in RY2 roots, with many Pi starvation regulated genes associated with P metabolic process, protein modification process, transport and other metabolic processes. A group of differentially expressed genes (DEGs) involved in Pi uptake and Pi homeostasis were identified, such as genes encoding Pi transporter (PT), purple acid phosphatase (PAP), and multidrug and toxin extrusion (MATE). Furthermore, a variety of genes related to transcription factors and regulators involved in Pi signaling, including genes belonging to the PHOSPHATE STARVATION RESPONSE 1-like (PHR1), WRKY and the SYG1/PHO81/XPR1 (SPX) domain, were also regulated by P deficiency in stylo roots.
Conclusions
This study reveals the possible mechanisms underlying the adaptation of stylo to P deficiency. The low-P tolerance in stylo is probably manifested through regulation of root growth, Pi acquisition and cellular Pi homeostasis as well as Pi signaling pathway. The identified genes involved in low-P tolerance can be potentially used to design the breeding strategy for develo** P-efficient stylo cultivars to grow on acid soils in the tropics.
Similar content being viewed by others


Background
Phosphorus (P) is one of the essential macronutrients for plant growth and development. P is involved in the processes of photosynthesis, respiration, energy metabolism and signal transduction in plants [1]. Furthermore, P is also an important structural component of various biomolecules in plant cells, including adenosine triphosphate (ATP), phospholipids, DNA and RNA [2]. Although total P is abundant in soils, P is easily immobilized by soil components into unavailable forms that cannot be directly utilized by plants [3]. Thus, low phosphate (Pi) availability is considered a major limiting factor for crop growth, especially in acid soils that occupy about 50% of the world’s arable land [3, 4]. To obtain high yields in traditional agricultural systems, farmers need to apply excessive quantities of P fertilizer, potentially leading to soil deterioration and eutrophication problems [5]. Furthermore, P fertilizer is derived from mined phosphate rock, which is a finite resource that is slowly depleting [6]. Therefore, improving the absorption and utilization of soil P can be an effective way for increasing crop yield and reducing fertilizer P application. Such improvements aim for the development of a more sustainable and environmentally sound agriculture.
To cope with low-P stress, plants have improved Pi uptake and homeostasis through a wide range of morphological, physiological and molecular changes, such as modifying root morphology and architecture, increasing secretion of organic acid and acid phosphatases, enhancing expression of high-affinity Pi transporters, develo** symbioses with arbuscular mycorrhizal fungi, and regulating complex P signaling networks in plant cells [7, 8]. It has been well demonstrated that plants display plasticity in root growth under P deficiency by changing root morphology and architecture, and thus increasing acquisition of P from the soil [9, 10]. For example, increase in root length and root/shoot ratio is observed in maize (Zea mays) [11], faba bean (Vicia faba) [12], rapeseed (Brassica napus) and wheat (Triticum aestivum) [13] in response to low P supply. Furthermore, acid phosphatase activities are up-regulated by Pi deprivation in many plants, such as rice (Oryza sativa), soybean (Glycine max) and chickpea (Cicer arietinum), which could contribute to increased organic P utilization [14,15,16].
To date, a variety of P responsive genes have been identified to participate in Pi uptake and homeostasis [8, 17]. For example, PHOSPHATE STARVATION RESPONSE 1 (AtPHR1) in Arabidopsis and OsPHR2 in rice, encoding the MYB transcription factor, are the central regulators involved in Pi signaling pathway [8]. AtPHR1 is demonstrated to regulate a set of Pi starvation induced (PSI) genes through binding to the P1BS cis element of target genes [18]. Furthermore, a negative regulatory role for protein containing the SYG1/PHO81/XPR1 (SPX) domain in rice is documented where OsSPX1 suppresses the transcripts of several PSI genes, such as Pi transporters (OsPT2 and OsPT6) and purple acid phosphatases (OsPAP10) [7]. A group of Pi transporters have been functionally characterized to be involved in Pi uptake and/or translocation in many plants; examples include: AtPT1 and AtPT2 from Arabidopsis [19, 20], OsPT1/9/10 from rice [21, 22] and GmPT5/7 from soybean [23, 24]. In addition, numerous purple acid phosphatase (PAP) homologues have also been demonstrated to function in Pi release from organic P, including AtPAP10/12/26 from Arabidopsis [25], OsPHY1 from rice [ In this study, two stylo (Stylosanthes guianensis) cultivars, ‘RY2’ and ‘RY5’, were used, which were widely grown in South China [33]. The stylo seeds were provided by the Institute of Tropical Crop Genetic Resources (TCGRI), Chinese Academy of Tropical Agricultural Sciences (CATAS), Hainan, China. Experiments were performed in a greenhouse at temperatures of 25 °C to 32 °C under natural sunlight with a photoperiod of about 13 h at the TCGRI, CATAS, Hainan, China (19°30′N, 109°30′E). Seeds were germinated for 3 d, and stylo seedlings were then transferred to a modified Hoagland nutrient solution containing 250 μM KH2PO4 for 14 d as previously described [71]. After that, seedlings were separately transplanted into nutrient solution supplied with 0, 100 and 250 μM KH2PO4, which were regarded as low (Pi deprivation), moderate and high P supply treatments, respectively. The nutrient solution was adjusted to a pH value of 5.8 and refreshed weekly. After 21 d of P treatments, shoots and roots were separately harvested for further analysis. An individual hydroponic box containing three seedlings of each stylo cultivar was set as one biological replicate. Each treatment included three biological replicates. Plant fresh roots were scanned using an Epson 12000XL scanner (Epson, Japan) with a resolution of 300 dpi, and the obtained image was saved as JEPG format. Total root length, root surface area and root volume were analyzed with WinRhizo Pro software (Regent Instruments Inc., Quebec, Canada). After that, shoot and root samples were oven dried at 75 °C for 7 d, and the dry mass of shoots and roots was further determined. For P concentration analysis, approximately 0.07 g dry samples were burned to ash at 600 °C in a muffle furnace. The sample of ash was absolutely dissolved in 100 mM HCl, and the supernatant was then used for P concentration analysis as previously described [72]. APase activities in stylo leaf and root were analyzed as previously described [38] with some modification. Approximately 0.15 g of leaf and root samples were ground in 1.5 mL of 45 mM Na-acetate buffer (pH 5.0) at 4 °C. After centrifugation at 12,000 rpm for 15 min at 4 °C, the supernatants were mixed with 2 mL of 45 mM Na-acetate buffer (pH 5.0) containing 1 mM ρ-nitrophenyl phosphate (Sigma, Saint Louis, MO, USA). After incubation at 37 °C for 15 min, the reaction was terminated by the addition of 1 mL of 1 M NaOH. APase activity was spectrophotometrically detected at 405 nm and expressed as micromoles of ρ-nitrophenyl phosphate hydrolyzed per mg protein per min. Protein concentration in the extracts was analyzed using the Coomassie Brilliant Blue staining method [73]. Total RNA from roots of RY2 at 0 (LP) and 250 (HP) μM KH2PO4 treatments was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. RNA purity and integrity were assessed by Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and Agilent 2100 (Agilent Technologies, Palo Alto, CA, USA), respectively. RNA sequencing analysis was conducted by Novogene Bioinformatics Technology Co., Ltd. (Bei**g, China). RNA-seq libraries were constructed using the NEBNext® UltraTM RNA Library Prep kit (NEB, Beverly, MA, USA), and the cDNA libraries were sequenced using an Illumina HiseqTM platform (Illumina, San Diego, CA, USA). The 150-bp paired-end reads (PE150) were generated. RNA-seq raw data were obtained using the Casava v.1.8 program. The raw reads in FASTQ format were processed, and then the high-quality clean reads were obtained after removing adaptor, ploy-N and low-quality sequences. The final clean reads were assembled using Trinity software (version 2). For annotation, all assembled unigenes were searched against a number of public databases, including the National Center for Biotechnology Information (NCBI) non-redundant protein sequences (Nr), the non-redundant nucleotide sequences (Nt), the Protein Family (Pfam), the Clusters of Orthologous Groups of protein database (COG), the Swiss-Prot protein database, the GO and the KEGG databases. The expression level of each gene was analyzed and represented by the expected number of fragments per kilobase of transcript sequence per millions base pairs (FPKM) using RSEM software with default settings [74]. Differentially expressed genes between two P treatments were identified using DESeq2 [75]. Genes with q-value <0.05 and |log2(fold change)| ≥1 were assigned as differentially expressed. GO and KEGG enrichment analyses of DEGs were performed as previously described [71, 76]. The interaction networks were analyzed by Cytoscape (version 3.8.0). The raw data were deposited in the Gene Expression Omnibus under GEO series number GSE171448. A total of 13 DEGs were selected to assess the accuracy of RNA-seq data using qRT-PCR method. qRT-PCR analysis was performed according to SYBR Green Master Mix kit (Vazyme, China), and was monitored on a QuantStudio™ 6 Flex Real-Time System (Thermo Fisher Scientific, Waltham, MA, USA). Specific primers of the tested genes are listed in Additional file 12. The relative expression of candidate gene was calculated relative to the expression of reference gene SgEF-1a as previously described [37]. Three biological replicates were included in the qRT-PCR analysis. Data analysis was performed for the mean and standard error calculation using Microsoft Excel 2003 (Microsoft Company, USA). One-way ANOVA and Student’s t-test analyses were performed with the SPSS program (SPSS Institute, USA, v. 13.0).Methods
Plant growth and treatments
Determination of root morphology and P concentration
Analysis of APase activity
RNA extraction and sequencing
Transcriptomic analysis
Validation of DEGs by qRT-PCR analysis
Statistical analysis
Availability of data and materials
The datasets are included in this article and its Additional files are available from the corresponding author on reasonable request. The datasets for this study can be found in the NCBI Gene Expression Omnibus under GEO series number of GSE171448.
Abbreviations
- ACP:
-
Acid phosphatase
- ALMT:
-
Aluminum-activated malate transporter
- ATP:
-
Adenosine triphosphate
- BP:
-
Biological processes
- CC:
-
Cellular components
- COG:
-
Clusters of orthologous groups of protein database
- DEGs:
-
Differentially expressed genes
- dNTP:
-
Deoxynucleoside triphosphate
- FPKM:
-
Expected number of fragments per kilobase of transcript sequence per millions base pairs
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MATE:
-
Multidrug and toxin extrusion
- MF:
-
Molecular function
- NCBI:
-
National center for biotechnology information
- Nr:
-
Non-redundant protein sequences
- Nt:
-
Non-redundant nucleotide sequences
- PAP:
-
Purple acid phosphatase
- Pfam:
-
Protein family
- PHR1:
-
PHOSPHATE STARVATION RESPONSE 1
- PPCK:
-
Phosphoenolpyruvate carboxylase kinase
- PSI:
-
Pi starvation induced
- PSR:
-
Pi starvation responses
- PT:
-
Pi transporter
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- RNA-seq:
-
RNA sequencing
- RNS:
-
Ribonuclease
- SPX:
-
SYG1/PHO81/XPR1
- TFs:
-
Transcription factors
References
Raghothama KG. Phosphate acquisition. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:665–93.
Mora-Macías J, Ojeda-Rivera JO, Gutiérrez-Alanís D, Yong-Villalobos L, Oropeza-Aburto A, Raya-González J, et al. Malate-dependent Fe accumulation is a critical checkpoint in the root developmental response to low phosphate. Proc Natl Acad Sci U S A. 2017;114(17):3563–72.
Ao JH, Chen ZJ, Wu M, Lu X, Huang Z, Liao H. Phosphorus fractions of red soils in Guangdong province of South China and their bioavailability for five crop species. Soil Sci. 2014;179:514–21.
Kochian LV, Piñeros MA, Liu J, Magalhaes JV. Plant adaptation to acid soils: The molecular basis for crop aluminum resistance. Annu Rev Plant Biol. 2015;66:571–98.
Conley DJ, Paerl HW, Howarth RW, Boesch DF, Seitzinger SP, Havens KE, et al. Controlling eutrophication: nitrogen and phosphorus. Science. 2009;323(5917):1014–5.
Cordell D, Drangert J, White S. The story of phosphorus: global food security and food for thought. Glob Environ Chang. 2009;19:292–305.
Wu P, Shou H, Xu G, Lian X. Improvement of phosphorus efficiency in rice on the basis of understanding phosphate signaling and homeostasis. Curr Opin Plant Biol. 2013;16(2):205–12.
Ham BK, Chen J, Yan Y, Lucas WJ. Insights into plant phosphate sensing and signaling. Curr Opin Biotechnol. 2018;49:1–9.
Lynch JP. Root phenes for enhanced soil exploration and phosphorus acquisition: tools for future crops. Plant Physiol. 2011;156(3):1041–9.
Rao IM, Miles JW, Beebe SE, Horst WJ. Root adaptations to soils with low fertility and aluminium toxicity. Ann Bot. 2016;118(4):593–605.
Bayuelo-Jiménez JS, Gallardo-Valdéz M, Pérez-Decelis VA, Magdaleno-Armas L, Ochoa I, Lynch JP. Genotypic variation for root traits of maize (Zea mays L.) from the Purhepecha Plateau under contrasting phosphorus availability. Field Crop Res. 2011;121(3):350–62.
Liu HT, White PJ, Li CJ. Biomass partitioning and rhizosphere responses of maize and faba bean to phosphorus deficiency. Crop Pasture Sci. 2016;67:847–56.
Lyu Y, Tang H, Li H, Zhang F, Rengel Z, Whalley WR, et al. Major crop species show differential balance between root morphological and physiological responses to variable phosphorus supply. Front Plant Sci. 2016;7:1939.
Mehra P, Pandey BK, Giri J. Improvement in phosphate acquisition and utilization by a secretory purple acid phosphatase (OsPAP21b) in rice. Plant Biotechnol J. 2017;15(8):1054–67.
Bhadouria J, Singh AP, Mehra P, Verma L, Srivastawa R, Parida SK, et al. Identification of purple acid phosphatases in chickpea and potential roles of CaPAP7 in seed phytate accumulation. Sci Rep. 2017;7(1):11012.
Zhu S, Chen M, Liang C, Xue Y, Lin S, Tian J. Characterization of purple acid phosphatase family and functional analysis of GmPAP7a/7b involved in extracellular ATP utilization in soybean. Front Plant Sci. 2020;11:661.
Liang C, Wang J, Zhao J, Tian J, Liao H. Control of phosphate homeostasis through gene regulation in crops. Curr Opin Plant Biol. 2014;21:59–66.
Chiou TJ, Lin SI. Signaling network in sensing phosphate availability in plants. Annu Rev Plant Biol. 2011;62:185–206.
Mitsukawa N, Okumura S, Shirano Y, Sato S, Kato T, Harashima S, et al. Overexpression of an Arabidopsis thaliana high-affinity phosphate transporter gene in tobacco cultured cells enhances cell growth under phosphate-limited conditions. Proc Natl Acad Sci U S A. 1997;94(13):7098–102.
Mudge SR, Rae AL, Diatloff E, Smith FW. Expression analysis suggests novel roles for members of the Pht1 family of phosphate transporters in Arabidopsis. Plant J. 2002;31(3):341–53.
Sun S, Gu M, Cao Y, Huang X, Zhang X, Ai P, et al. A constitutive expressed phosphate transporter, OsPht1;1, modulates phosphate uptake and translocation in phosphate-replete rice. Plant Physiol. 2012;159(4):1571–81.
Wang X, Wang Y, Piñeros MA, Wang Z, Wang W, Li C, et al. Phosphate transporters OsPHT1;9 and OsPHT1;10 are involved in phosphate uptake in rice. Plant Cell Environ. 2014;37(5):1159–70.
Qin L, Zhao J, Tian J, Chen L, Sun Z, Guo Y, et al. The high-affinity phosphate transporter GmPT5 regulates phosphate transport to nodules and nodulation in soybean. Plant Physiol. 2012;159(4):1634–43.
Chen L, Qin L, Zhou L, Li X, Chen Z, Sun L, et al. A nodule-localized phosphate transporter GmPT7 plays an important role in enhancing symbiotic N2 fixation and yield in soybean. New Phytol. 2019;221(4):2013–25.
Wang L, Lu S, Zhang Y, Li Z, Du X, Liu D. Comparative genetic analysis of Arabidopsis purple acid phosphatases AtPAP10, AtPAP12, and AtPAP26 provides new insights into their roles in plant adaptation to phosphate deprivation. J Integr Plant Biol. 2014;56(3):299–314.
Li RJ, Lu WJ, Guo CJ, Li XJ, Gu JT, **ao K. Molecular characterization and functional analysis of OsPHY1, a purple acid phosphatase (PAP)-type phytase gene in rice (Oryza sativa L.). J Integr Plant Biol. 2012;11:1217–26.
Liang C, Sun L, Yao Z, Liao H, Tian J. Comparative analysis of PvPAP gene family and their functions in response to phosphorus deficiency in common bean. PLoS One. 2012;7(5):e38106.
Kong Y, Li X, Wang B, Li W, Du H, Zhang C. The soybean purple acid phosphatase GmPAP14 predominantly enhances external phytate utilization in plants. Front Plant Sci. 2018;9:292.
Li C, Zhou J, Wang X, Liao H. A purple acid phosphatase, GmPAP33, participates in arbuscule degeneration during arbuscular mycorrhizal symbiosis in soybean. Plant Cell Environ. 2019;42(6):2015–27.
Guo W, Zhao J, Li X, Qin L, Yan X, Liao H. A soybean β-expansin gene GmEXPB2 intrinsically involved in root system architecture responses to abiotic stresses. Plant J. 2011;66(3):541–52.
Yu Z, Kang B, He X, Lv S, Bai Y, Ding W, et al. Root hair-specific expansins modulate root hair elongation in rice. Plant J. 2011;66(5):725–34.
Chandra A. Diversity among Stylosanthes species: habitat, edaphic and agro-climatic affinities leading to cultivar development. J Environ Biol. 2009;30(4):471–8.
Liu GD, Bai CJ, Wang DJ, He HX. Stylosanthes cultivars in China: their development and performance. In: Chakraborty S, editor. High-yielding anthracnose-resistant Stylosanthes for agricultural systems: Australian Centre for International Agricultural Research (ACIAR) Press; 2004. p. 156–8.
Guo PF, Liu PD, Lei J, Chen CH, Qiu H, Liu GD, et al. Improvement of plant regeneration and Agrobacterium-mediated genetic transformation of Stylosanthes guianensis. Trop Grassl Forrajes. 2019;7:480–92.
Sun L, Liang C, Chen Z, Liu P, Tian J, Liu G, et al. Superior aluminium (Al) tolerance of Stylosanthes is achieved mainly by malate synthesis through an Al-enhanced malic enzyme, SgME1. New Phytol. 2014;202(1):209–19.
Chen Z, Sun L, Liu P, Liu G, Tian J, Liao H. Malate synthesis and secretion mediated by a manganese-enhanced malate dehydrogenase confers superior manganese tolerance in Stylosanthes guianensis. Plant Physiol. 2015;167(1):176–88.
Liu P, Cai Z, Chen Z, Mo X, Ding X, Liang C, et al. A root-associated purple acid phosphatase, SgPAP23, mediates extracellular phytate-P utilization in Stylosanthes guianensis. Plant Cell Environ. 2018;41(12):2821–34.
Liu PD, Xue YB, Chen ZJ, Liu GD, Tian J. Characterization of purple acid phosphatases involved in extracellular dNTP utilization in Stylosanthes. J Exp Bot. 2016;67(14):4141–54.
Du YM, Tian J, Liao H, Bai CJ, Yan XL, Liu GD. Aluminium tolerance and high phosphorus efficiency helps Stylosanthes better adapt to low-P acid soils. Ann Bot. 2009;103(8):1239–47.
Gamuyao R, Chin JH, Pariasca-Tanaka J, Pesaresi P, Catausan S, Dalid C, et al. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature. 2012;488(7412):535–9.
Wu W, Lin Y, Liu P, Chen Q, Tian J, Liang C. Association of extracellular dNTP utilization with a GmPAP1-like protein identified in cell wall proteomic analysis of soybean roots. J Exp Bot. 2018;69(3):603–17.
Lin Y, Chen G, Hu H, Yang X, Zhang Z, Jiang X, et al. Phenotypic and genetic variation in phosphorus-deficiency-tolerance traits in Chinese wheat landraces. BMC Plant Biol. 2020;20(1):330.
Lan P, Li WF, Schmidt W. Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol Cell Proteomics. 2012;11(11):1156–66.
Secco D, Jabnoune M, Walker H, Shou H, Wu P, Poirier Y, et al. Spatio-temporal transcript profiling of rice roots and shoots in response to phosphate starvation and recovery. Plant Cell. 2013;25(11):4285–304.
Zeng HQ, Wang GP, Zhang YQ, Hu XY, Pi EX, Zhu YY, et al. Genome-wide identification of phosphate-deficiency-responsive genes in soybean roots by high-throughput sequencin. Plant Soil. 2016;398:207–27.
Feng H, Li B, Zhi Y, Chen J, Li R, **a X, et al. Overexpression of the nitrate transporter, OsNRT2.3b, improves rice phosphorus uptake and translocation. Plant Cell Rep. 2017;36(8):1287–96.
Liu F, Chang XJ, Ye Y, **e WB, Wu P, Lian XM. Comprehensive sequence and whole-life-cycle expression profile analysis of the phosphate transporter gene family in rice. Mol Plant. 2011;4(6):1105–22.
Cao Y, Liu J, Li Y, Zhang J, Li S, An Y, et al. Functional analysis of the phosphate transporter gene MtPT6 from Medicago truncatula. Front Plant Sci. 2021;11:620377.
Karthikeyan AS, Varadarajan DK, Mukatira UT, D'Urzo MP, Damsz B, Raghothama KG. Regulated expression of Arabidopsis phosphate transporters. Plant Physiol. 2002;130(1):221–33.
Shin H, Shin HS, Dewbre GR, Harrison MJ. Phosphate transport in Arabidopsis: Pht1;1 and Pht1;4 play a major role in phosphate acquisition from both low- and high-phosphate environments. Plant J. 2004;39(4):629–42.
Ai P, Sun S, Zhao J, Fan X, **n W, Guo Q, et al. Two rice phosphate transporters, OsPht1;2 and OsPht1;6, have different functions and kinetic properties in uptake and translocation. Plant J. 2009;57(5):798–809.
Peng W, Wu W, Peng J, Li J, Lin Y, Wang Y, et al. Characterization of the soybean GmALMT family genes and the function of GmALMT5 in response to phosphate starvation. J Integr Plant Biol. 2018;60(3):216–31.
Magalhaes JV, Liu J, Guimarães CT, Lana UG, Alves VM, Wang YH, et al. A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat Genet. 2007;39(9):1156–61.
Liang C, Piñeros MA, Tian J, Yao Z, Sun L, Liu J, et al. Low pH, aluminum, and phosphorus coordinately regulate malate exudation through GmALMT1 to improve soybean adaptation to acid soils. Plant Physiol. 2013;161(3):1347–61.
Chen Q, Wang L, Liu D, Ma S, Dai Y, Zhang X, et al. Identification and expression of the multidrug and toxic compound extrusion (MATE) gene family in Capsicum annuum and Solanum tuberosum. Plants. 2020;9(11):1448.
Li XF, Zuo FH, Ling GZ, Li YY, Yu YX, Yang PQ, et al. Secretion of citrate from roots in response to aluminum and low phosphorus stresses in Stylosanthes. Plant Soil. 2009;325:219–29.
Haran S, Logendra S, Seskar M, Bratanova M, Raskin I. Characterization of Arabidopsis acid phosphatase promoter and regulation of acid phosphatase expression. Plant Physiol. 2000;124(2):615–26.
Zhang Q, Wang C, Tian J, Li K, Shou H. Identification of rice purple acid phosphatases related to phosphate starvation signalling. Plant Biol. 2011;13(1):7–15.
Li C, Gui S, Yang T, Walk T, Wang X, Liao H. Identification of soybean purple acid phosphatase genes and their expression responses to phosphorus availability and symbiosis. Ann Bot. 2012;109(1):275–85.
Luo J, Liu Y, Zhang H, Wang J, Chen Z, Luo L, et al. Metabolic alterations provide insights into Stylosanthes roots responding to phosphorus deficiency. BMC Plant Biol. 2020;20(1):85.
Valdés-López O, Arenas-Huertero C, Ramírez M, Girard L, Sánchez F, Vance CP, et al. Essential role of MYB transcription factor: PvPHR1 and microRNA: PvmiR399 in phosphorus-deficiency signalling in common bean roots. Plant Cell Environ. 2008;31(12):1834–43.
Guo M, Ruan W, Li C, Huang F, Zeng M, Liu Y, et al. Integrative comparison of the role of the PHOSPHATE RESPONSE1 subfamily in phosphate signaling and homeostasis in rice. Plant Physiol. 2015;168(4):1762–76.
Sun L, Song L, Zhang Y, Zheng Z, Liu D. Arabidopsis PHL2 and PHR1 act redundantly as the key components of the central regulatory system controlling transcriptional responses to phosphate starvation. Plant Physiol. 2016;170(1):499–514.
Xue YB, **ao BX, Zhu SN, Mo XH, Liang CY, Tian J, et al. GmPHR25, a GmPHR member up-regulated by phosphate starvation, controls phosphate homeostasis in soybean. J Exp Bot. 2017;68(17):4951–67.
Chen YF, Li LQ, Xu Q, Kong YH, Wang H, Wu WH. The WRKY6 transcription factor modulates PHOSPHATE1 expression in response to low Pi stress in Arabidopsis. Plant Cell. 2009;21(11):3554–66.
Devaiah BN, Karthikeyan AS, Raghothama KG. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007;143(4):1789–801.
Yao Z, Tian J, Liao H. Comparative characterization of GmSPX members reveals that GmSPX3 is involved in phosphate homeostasis in soybean. Ann Bot. 2014;114(3):477–88.
Duan K, Yi K, Dang L, Huang H, Wu W, Wu P. Characterization of a sub-family of Arabidopsis genes with the SPX domain reveals their diverse functions in plant tolerance to phosphorus starvation. Plant J. 2008;54(6):965–75.
Puga MI, Mateos I, Charukesi R, Wang Z, Franco-Zorrilla JM, de Lorenzo L, et al. SPX1 is a phosphate-dependent inhibitor of PHOSPHATEF STARVATION RESPONSE 1 in Arabidopsis. Proc Natl Acad Sci U S A. 2014;111(41):14947–52.
Lv Q, Zhong Y, Wang Y, Wang Z, Zhang L, Shi J, et al. SPX4 negatively regulates phosphate signaling and homeostasis through its interaction with PHR2 in rice. Plant Cell. 2014;26(4):1586–97.
Jia Y, Li X, Liu Q, Hu X, Li J, Dong R, et al. Physiological and transcriptomic analyses reveal the roles of secondary metabolism in the adaptive responses of Stylosanthes to manganese toxicity. BMC Genomics. 2020;21(1):861.
Murphy J, Riley JP. A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta. 1962;27:31–6.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Acknowledgements
We thank **aoyan Zou for assistance in preparation of figures and Chun Liu for providing critical comments about this work.
Funding
The research was supported by the National Natural Science Foundation of China (31801951, 31861143013, 31802125), the Modern Agro-industry Technology Research System (CARS-34), and the Integrated Demonstration of Key Techniques for the Industrial Development of Featured Crops in Rocky Desertification Areas of Yunnan-Guangxi-Guizhou Provinces (SMH2019-2021). The authors declare that none of the funding bodies have any role in the design of the study or collection, analysis, and interpretation of data as well as in writing the manuscript.
Author information
Authors and Affiliations
Contributions
X.M. and G.L. designed the research. Z.C., J.S. and X.L. performed the physiological experiments. Z.C. J.A. and J.A.C. carried out the transcriptomic analysis. Z.C., X.M., G.L., I.R., R.S.K., M.P. and X.M. analyzed the data and wrote the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The stylo seeds used in this study were provided by the Tropical Pasture Research Center, Institute of Tropical Crop Genetic Resources (TCGRI), Chinese Academy of Tropical Agriculture Sciences (CATAS), Hainan, China. All experimental researches were performed in accordance with the institutional and national guidelines of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The decrease rate of growth parameters of stylo under 0 (LP) and 100 μM KH2PO4 relative to that in 250 (HP) μM KH2PO4 treatment.
Additional file 2: Table S2.
Summary of stylo roots transcriptomes in 0 (LP) and 250 (HP) μM KH2PO4 supply treatments.
Additional file 3: Table S3.
DEGs identified in stylo roots under 0 (LP) and 250 (HP) μM KH2PO4 supply treatments.
Additional file 4: Figure S1.
Clustering analysis of the DEGs under two P conditions. The transcripts of DEGs were normalized as log2(FPKM). Expression levels ranged from red to blue indicate high to low expression for genes, respectively. LP and HP represent 0 and 250 μM KH2PO4 supply treatments, respectively.
Additional file 5: Table S4.
DEGs related to transporters.
Additional file 6: Table S5.
DEGs encoding putative phosphatases.
Additional file 7: Table S6.
DEGs related to root growth.
Additional file 8: Table S7.
Potential transcription factors involved in Pi signaling.
Additional file 9: Table S8
. DEGs related to plant hormone signal transduction.
Additional file 10: Figure S2.
Heatmap analysis of the DEGs belonging to SPX containing proteins. The transcripts of DEGs were normalized as log2(FPKM+1). Gene IDs were showed by the legend on the right. Expression levels ranged from red to blue indicate high to low expression for genes, respectively. LP and HP represent 0 and 250 μM KH2PO4 supply treatments, respectively.
Additional file 11: Figure S3.
Correlation analysis of gene expression between transcriptome data and qRT-PCR results. Nine up-regulated and four down-regulated DEGs were selected for qRT-PCR analysis. Transcriptome data were plotted against data from qRT-PCR. Data are presented on a log2 scale.
Additional file 12: Table S9.
Primers used for qRT-PCR analysis of the selected genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Z., Song, J., Li, X. et al. Physiological responses and transcriptomic changes reveal the mechanisms underlying adaptation of Stylosanthes guianensis to phosphorus deficiency. BMC Plant Biol 21, 466 (2021). https://doi.org/10.1186/s12870-021-03249-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03249-2