
Abstract
Background
Two-component systems (TCSs) play a crucial role in the growth of Mycobacterium tuberculosis (M. tuberculosis). However, the precise regulatory mechanism of their contribution remain to be elucidated, and only a limited number of studies have investigated the impact of gene mutations within TCSs on the transmission of M. tuberculosis. Therefore, this study aims to explore the relationship between TCSs gene mutation and the global transmission of M. tuberculosis.
Results
A total of 13531 M.tuberculosis strains were enrolled in the study. Most of the M.tuberculosis strains belonged to lineage4 (n=6497,48.0%), followed by lineage2 (n=5136,38.0%). Our results showed that a total of 36 single nucleotide polymorphisms (SNPs) were positively correlated with clustering of lineage2, such as Rv0758 (phoR, C820G), Rv1747(T1102C), and Rv1057(C1168T). A total of 30 SNPs showed positive correlation with clustering of lineage4, such as phoR(C182A, C1184G, C662T, T758G), Rv3764c (tcrY, G1151T), and Rv1747 C20T. A total of 19 SNPs were positively correlated with cross-country transmission of lineage2, such as phoR A575C, Rv1028c (kdpD, G383T, G1246C), and Rv1057 G817T. A total of 41 SNPs were positively correlated with cross-country transmission of lineage4, such as phoR(T758G, T327G, C284G), kdpD(G1755A, G625C), Rv1057 C980T, and Rv1747 T373G.
Conclusions
Our study identified that SNPs in genes of two-component systems were related to the transmission of M. tuberculosis. This finding adds another layer of complexity to M. tuberculosis virulence and provides insight into future research that will help to elucidate a novel mechanism of M. tuberculosis pathogenicity.
Similar content being viewed by others
Background
Tuberculosis is a serious global health problem caused by Mycobacterium tuberculosis (M. tuberculosis), a pathogen that lives and thrives inside human cells [1]. It is a highly contagious and often fatal disease that affects millions of people worldwide, making it a significant burden on public health systems and societies. However, despite its enormous global burden, the factors that contribute to tuberculosis transmission are still poorly understood. Therefore, develo** a better understanding of M. tuberculosis transmission is critical for guiding effective tuberculosis control strategies and reducing the disease’s burden on society.
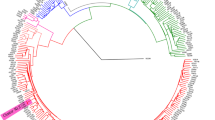
Bacterial two-component systems (TCSs) are the most important sensing mechanisms that respond to a diverse range of ligands, including ions, gases, and metabolites. In pathogenic bacteria, TCSs play a crucial role in promoting pathogenesis by regulating bacterial gene expression in response to hostile host environments or metabolic stresses [2, 37](Additional file 2: Tables S1-S2). Construction of the maximum likelihood phylogenetic tree was conducted through the IQ-TREE software package (version 1.6.12), utilizing the JC nucleotide substitution model and gamma model of rate heterogeneity, with 100 bootstrap replicates included [38]. Mycobacterium canettii CIPT140010059 was deemed to be an outlier. The resultant phylogenetic tree was visualized through the utilization of iTOL (https://itol.embl.de/) (Fig. 3, Additional file 1: Figs. S1–S7).

The phylogenetic tree analysis of lineage2.2. (A) the phylogenetic tree analysis of lineage2.2.1. (B) the phylogenetic tree analysis of lineage2.2.2
Propagation analysis
Cluster analysis was utilized to investigate the influence of two-component system gene mutations on the transmission of M. tuberculosis [39]. Based on a previous study [40], we applied clustering to define transmission clusters and used a threshold of less than 25 SNPs. In addition, we chose the threshold of 25 SNPs because our isolates were spread in terms of location and time (1991–2019) and because we were probably missing several intermediary isolates (and cases) in our collection. (Additional file 2: Tables S1-S2). Additionally, according to the classification of transmission clusters by scholars, we also divided transmission clusters into large, medium, or small (large, over 75th percentile; medium, between 25th and 75th percentile; and small, under 25th percentile) [14]. To enhance understanding of the global distribution patterns and conduct an extensive analysis of the transmission dynamics of M.tuberculosis strains, we classified them into cross-country and within-country clusters. Furthermore, we categorized the M. tuberculosis strains into cross-regional and within-regional clusters based on geographic location utilizing the United Nations standard regions (UN M.49).
Acquisition of two-component system genes
A total of 45 two-component system genes were obtained according to NCBI and literature search [2, 7, 41]. Python was utilized to detect mutations in genes associated with TCSs (Additional file 2: Table S3).
Modeling and statistical analysis
Prediction models including gradient boosting decision tree and random forest were established by machine learning using the Scikit-learn Python package. We randomly divided all samples into training and test sets at a ratio of 7:3. Each of the models was evaluated with the metrics of Kappa, sensitivity, specificity, accuracy, positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR) and area under curve (AUC) [42]. After the model was fitted, we evaluated the importance of the input variables on the model. To enhance the precision of predicting risk factors, we utilized the score to assess the influence of each input feature of the models, and take the intersection of both conditions and obtain the top-performing accessions as the important features [43, 44]. Subsequently, we established the generalized linear mixed model by using the statsmodels.api Python package to further analyze the important features and obtain the final influencing factors. All statistical analyses were performed using SPSS 26.0. All statistical tests were two-tailed, and P values less than 0.05 were considered statistically significant.
Data Availability
The whole genome sequences have been submitted to the NCBI under the accession number PRJNA1002108.
Change history
08 January 2024
A Correction to this paper has been published: https://doi.org/10.1186/s12864-023-09914-0
Abbreviations
- M. tuberculosis :
-
Mycobacterium tuberculosis
- TCSs:
-
Two-component systems
- WGS:
-
Whole genome sequencing
- SPHCC:
-
Shandong Public Health Clinical Research Center
- WRCH:
-
Weifang Respiratory Clinical Hospital
- CTAB:
-
Cetyltrimethylammonium Bromide
- QC:
-
Quality control
- SNP:
-
Single nucleotide polymorphism
- SNPs:
-
Single nucleotide polymorphisms
- NCBI:
-
National Center for Biotechnology Information
- PPV:
-
Positive Predictive Value
- NPV:
-
Negative Predictive Value
- PLR:
-
Positive Likelihood Ratio
- NLR:
-
Negative Likelihood Ratio
- AUC:
-
Area Under Curve
References
World Health Organization. Global Tuberculosis report 2022. Geneva: World Health Organization; 2022.
Parish T. Two-Component Regulatory Systems of Mycobacteria. Microbiol Spectr. 2014;2(1):MGM2-0010-2013.
Zhou PF, Long QX, Zhou YX, Wang HH, **e J. Mycobacterium tuberculosis two-Component systems and implications in novel vaccines and Drugs. Crit Rev Eukar Gene Expr. 2012;22:37–52.
Kusebauch U, Ortega C, Ollodart A, Rogers RS, Sherman DR, Moritz RL, et al. Mycobacterium tuberculosis supports protein tyrosine phosphorylation. Proc Natl Acad Sci U S A. 2014;111:9265–70.
Buglino JA, Sankhe GD, Lazar N, Bean JM, Glickman MS. Integrated sensing of host stresses by inhibition of a cytoplasmic two-component system controls M. Tuberculosis acute lung Infection. eLife. 2021;10:e65351.
Supply P, Magdalena J, Himpens S, Locht C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997;26:991–1003.
Bretl DJ, Demetriadou C, Zahrt TC. Adaptation to Environmental Stimuli within the host: Two-Component Signal Transduction Systems of Mycobacterium tuberculosis. Microbiol Mol Biol Rev. 2011;75:566–82.
Takiff HE, Feo O. Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect Dis. 2015;15:1077–90.
Walker TM, Kohl TA, Omar SV, Hedge J, Del Ojo Elias C, Bradley P, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15:1193–202.
Köser CU, Bryant JM, Becq J, Török ME, Ellington MJ, Marti-Renom MA, et al. Whole-genome sequencing for rapid susceptibility testing of M. Tuberculosis. N Engl J Med. 2013;369:290–2.
Waturuocha UW, Krishna MS, Malhotra V, Dixit NM, Saini DK. A low-prevalence single-nucleotide polymorphism in the Sensor kinase PhoR in Mycobacterium tuberculosis suppresses its autophosphatase activity and reduces pathogenic fitness: implications in Evolutionary Selection. Front Microbiol. 2021;12:724482.
Mendes MV, Tunca S, Antón N, Recio E, Sola-Landa A, Aparicio JF, et al. The two-component phor-phop system of Streptomyces natalensis: inactivation or deletion of phoP reduces the negative phosphate regulation of pimaricin biosynthesis. Metab Eng. 2007;9:217–27.
De Maio F, Berisio R, Manganelli R, Delogu G. PE_PGRS proteins of Mycobacterium tuberculosis: a specialized molecular task force at the forefront of host-pathogen interaction. Virulence. 2020;11:898–915.
Chiner-Oms Á, Sánchez-Busó L, Corander J, Gagneux S, Harris SR, Young D et al. Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex. Sci Adv. 2019.
Vashist A, Malhotra V, Sharma G, Tyagi JS, Clark-Curtiss JE. Interplay of PhoP and DevR response regulators defines expression of the dormancy regulon in virulent Mycobacterium tuberculosis. J Biol Chem. 2018;293:16413–25.
Ryndak M, Wang S, Smith I. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 2008;16:528–34.
Agrawal R, Saini DK. Rv1027c-Rv1028c encode functional KdpDE two–component system in Mycobacterium tuberculosis. Biochem Biophys Res Commun. 2014;446:1172–8.
Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84.
Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during Infection. Proc Natl Acad Sci U S A. 2003;100:12989–94.
Parish T, Smith DA, Kendall S, Casali N, Bancroft GJ, Stoker NG. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect Immun. 2003;71:1134–40.
Haydel SE, Benjamin WH, Dunlap NE, Clark-Curtiss JE. Expression, autoregulation, and DNA binding properties of the Mycobacterium tuberculosis TrcR response regulator. J Bacteriol. 2002;184:2192–203.
Wernisch L, Kendall SL, Soneji S, Wietzorrek A, Parish T, Hinds J, et al. Analysis of whole-genome microarray replicates using mixed models. Bioinformatics. 2003;19:53–61.
Haydel SE, Dunlap NE, Benjamin WH. In vitro evidence of two-component system phosphorylation between the Mycobacterium tuberculosis TrcR/TrcS proteins. Microb Pathog. 1999;26:195–206.
Chen X, He G, Wang S, Lin S, Chen J, Zhang W. Evaluation of whole-genome sequence method to Diagnose Resistance of 13 anti-tuberculosis Drugs and characterize resistance genes in clinical Multi-drug Resistance Mycobacterium tuberculosis isolates from China. Front Microbiol. 2019;10:1741.
Yang C, Luo T, Shen X, Wu J, Gan M, Xu P, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017;17:275–84.
Koster KJ, Largen A, Foster JT, Drees KP, Qian L, Desmond E, et al. Genomic sequencing is required for identification of Tuberculosis transmission in Hawaii. BMC Infect Dis. 2018;18:608.
Hicks ND, Yang J, Zhang X, Zhao B, Grad YH, Liu L, et al. Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat Microbiol. 2018;3:1032–42.
Liu Q, Ma A, Wei L, Pang Y, Wu B, Luo T, et al. China’s Tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol. 2018;2:1982–92.
Huang H, Ding N, Yang T, Li C, Jia X, Wang G, et al. Cross-sectional whole-genome sequencing and epidemiological study of Multidrug-resistant Mycobacterium tuberculosis in China. Clin Infect Dis. 2019;69:405–13.
Luo T, Comas I, Luo D, Lu B, Wu J, Wei L, et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Bei**g family with Han Chinese. Proc Natl Acad Sci USA. 2015;112:8136–41.
Jiang Q, Liu Q, Ji L, Li J, Zeng Y, Meng L, et al. Citywide transmission of Multidrug-resistant Tuberculosis under China’s Rapid Urbanization: a Retrospective Population-based genomic spatial epidemiological study. Clin Infect Dis. 2020;71:142–51.
Coll F, Phelan J, Hill-Cawthorne GA, Nair MB, Mallard K, Ali S, et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat Genet. 2018;50:307–16.
Jung Y, Han D, BWA-MEME. BWA-MEM emulated with a machine learning approach. Bioinformatics. 2022;:btac137.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Liu F, Zhang Y, Zhang L, Li Z, Fang Q, Gao R, et al. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019;20:242.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Coll F, McNerney R, Guerra-Assunção JA, Glynn JR, Perdigão J, Viveiros M, et al. A robust SNP barcode for ty** Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Seto J, Wada T, Suzuki Y, Ikeda T, Mizuta K, Yamamoto T, et al. Mycobacterium tuberculosis Transmission among Elderly persons, Yamagata Prefecture, Japan, 2009–2015. Emerg Infect Dis. 2017;23:448–55.
Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46.
Li X, Lv X, Lin Y, Zhen J, Ruan C, Duan W, et al. Role of two-component regulatory systems in intracellular survival of Mycobacterium tuberculosis. J Cell Biochem. 2019;120:12197–207.
Luo Y, Xue Y, Song H, Tang G, Liu W, Bai H, et al. Machine learning based on routine laboratory indicators promoting the discrimination between active Tuberculosis and latent Tuberculosis Infection. J Infect. 2022;84:648–57.
Bi X, Xu Q, Luo X, Sun Q, Wang Z. Weighted Random Support Vector machine clusters analysis of resting-state fMRI in mild cognitive impairment. Front Psychiatry. 2018;9:340.
Agarwal G, Saade S, Shahid M, Tester M, Sun Y. Quantile function modeling with application to salinity tolerance analysis of plant data. BMC Plant Biol. 2019;19:526.
Acknowledgements
We thank Shandong Provincial Hospital, Shandong Provincial Chest Hospital, 13 municipal-level and 21 county-level local health departments for drug susceptibility data, demographic, and clinical data.
Funding
This work was supported by the Department of Science & Technology of Shandong Province (CN) (No.2007GG30002033; No.2017GSF218052) and the **an Science and Technology Bureau (CN) (No.201704100). The funding body/bodies did not provide any assistance in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
HCL, YL, and YML participated in the study design. YL, HCL, YML, XLK, NNT, and YFL performed data collection and statistical analyses. YL, TTW, YYL, and YWH helped draft the manuscript. YWH, QLH, and YYL overviewed and supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study complies with the Declaration of Helsinki, and was approved by the Ethics Committee of Shandong Provincial Hospital, affiliated with Shandong University (SPH), the Ethics Weifang Respiratory Clinical Hospital (WRCH) and the Ethics Committee of Shandong Provincial Chest Hospital (SPCH), which waived informed patient consent because all patient records and information were anonymized and deidentified before the analysis.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: there was an error in affiliation 1 and the Background section of the Abstract had to be revised.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Kong, X., Li, Y. et al. Association between two-component systems gene mutation and Mycobacterium tuberculosis transmission revealed by whole genome sequencing. BMC Genomics 24, 718 (2023). https://doi.org/10.1186/s12864-023-09788-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09788-2