
Abstract
Background
Long non-coding RNAs (lncRNAs) are involved in many fundamental biological processes, such as transcription regulation, protein degradation, and cell differentiation. Information on lncRNA in the melon fly, Zeugodacus cucurbitae (Coquillett) is currently limited.
Results
We constructed 24 RNA-seq libraries from eight tissues (midgut, Malpighian tubules, fat body, ovary, and testis) of Z. cucurbitae adults. A total of 3124 lncRNA transcripts were identified. Among those, 1464 were lincRNAs, 1037 were intronic lncRNAs, 301 were anti-sense lncRNAs, and 322 were sense lncRNAs. The majority of lncRNAs contained two exons and one isoform. Differentially expressed lncRNAs were analyzed between tissues, and Malpighian tubules versus testis had the largest number. Some lncRNAs exhibited strong tissue specificity. Specifically expressed lncRNAs were identified and filtered in tissues of female and male Z. cucurbitae based on their expression levels. Four midgut-specific lncRNAs were validated by quantitative real-time polymerase chain reaction (RT-qPCR), and the data were consistent with RNA-seq data. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of targets of midgut-specific lncRNAs indicated an enrichment of the metabolic process.
Conclusions
This was the first systematic identification of lncRNA in the melon fly. Expressions of lncRNAs in multiple adult tissues were evaluated by quantitative transcriptomic analysis. These qualitative and quantitative analyses of lncRNAs, especially the tissue-specific lncRNAs in Z. cucurbitae, provide useful data for further functional studies.
Similar content being viewed by others


Background
The high-throughput sequencing technology has greatly stimulated studies of insect genomes and transcriptomes [11]. LncRNAs show poor conservation among different species and have relatively low expression level compared with mRNAs [12].
Systematic identification and analyses of lncRNAs have been investigated in various species, such as goat [13], mouse [14], zebrafish [15], tilapia [4], chicken [16], and fungus [17]. Many studies have provided data enabling lncRNA identification in insects. In Drosophila melanogaster, a total of 1875 candidate lncRNAs were identified from multiple transcriptome data sets [18]. Using RNA-seq technology, 8096 putative lncRNAs were identified in one susceptible and two insecticide-resistant strains of Plutella xylostella [19]. In addition, 2949 lncRNAs were found in RNA-seq data of multiple life stages of Anopheles gambiae [20]. These studies increased the catalog of insect lncRNAs and provided insight into their functions, such as cell differentiation, transcription regulation, and dosage compensation [1]. Compared with mRNA, lncRNA exhibits more tissue specific-expression in insects, indicating a specific function associated with these tissues [21].
LncRNAs can play crucial roles in many biological processes, such as cell differentiation and development [22, 23]. In Drosophila, lncRNAs were probable involved in molting because the mass of lncRNAs was significantly up-regulated in the late embryonic and larval stages [5]. Knockdown of lincRNA_1317 expression by RNA interference suppressed the replication of dengue virus in Aedes aegypti, demonstrating the essential role of the lncRNA in anti-viral defenses [24]. Genome location and co-expression analyses of protein-coding genes and lncRNAs revealed that several lncRNAs might be associated with fecundity and virulence in Nilaparvata lugens [1]. More interestingly, specific expression of lncRNAs among tissues suggested their associated functions. In Locusta migratoria, knockdown of a brain-specific lncRNA (PAHAL) by RNA interference reduced aggregation behavior [25]. Functional annotation of target genes of testis-specific lncRNAs from RNA-seq data indicated that they may participate in the spermatogenesis of Bombyx mori [26].
The melon fly, Zeugodacus cucurbitae (Coquillett), is one of the most destructive and troublesome agricultural pests [27, 28]. The genome of Z. cucurbitae has been sequenced and released [29], which provides sequence information for gene annotation and functional research. The genome-wide expression of genes during the developmental stages has also been analyzed by RNA-seq [30]. However, there is no information about lncRNAs or functional studies in Z. cucurbitae. In this study, 24 RNA-seq datasets were constructed from different tissues of female and male Z. cucurbitae, including midgut, Malpighian tubules, fat body, ovary, and testis. By the way, a total of 3124 lncRNAs were strictly identified from the RNA-seq data, and their features and characteristics were analyzed. Differentially expressed lncRNAs between tissues in female and male adults, as well as similar tissues in female and male adults, were analyzed. Tissue-specific lncRNAs were screened in female and male tissues based on their relative expression levels. GO and KEGG pathway enrichment analysis of targets of midgut-specific lncRNAs revealed unique functional annotations. Our findings create a catalog of lncRNAs in tissues of Z. cucurbitae and provide information that will be useful for further functional studies.
Results
Identification and characterization of lncRNAs
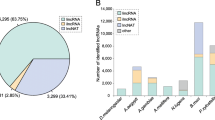
A total of 511,526,830 raw reads were generated from 24 RNA-seq datasets. Q30 scores were ≥ 93.0% in all of the samples. GC contents ranged from 40.1 to 46.69%. The accuracy of RNA-seq data was of high degrees as no “N” base was detected in any of the samples (Table 1). All of the RNA sequencing data produced in this study are available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under the accession number: PRJNA579200. After filtering under a computational pipeline (Fig. S1), a total of 22,159 lncRNA candidates were retained. Null-expressed transcripts (FPKM value < 1 in all analyzed samples) were discarded, and the numbers of lncRNAs in female and male tissues were screened. In females, the largest population of lncRNAs (1024) was found in the Malpighian tubules (Fig. 1a). There were 20,330 null-expressed lncRNAs in female tissues (Fig. 1b). Fat body had the largest lncRNA population (1026) among male tissues (Fig. 1c). Male tissues had 19,680 null-expressed lncRNAs (Fig. 1d). After discarding all null-expressed lncRNAs, a total of 3124 lncRNA transcripts were strictly identified from the transcriptome data of the eight tissues. Most of these were lincRNAs (1464; 46.9%), followed by intronic lncRNAs (1037; 33.2%), anti-sense lncRNAs (301; 9.6%), and sense lncRNA (322; 10.3%) (Fig. 2a). The lncRNA length distribution showed that most lncRNA transcripts were longer than 3000 nucleotides (Fig. 2b). The majority of lncRNAs only had one isoform (Fig. 2c). Most of the lncRNAs in Z. cucurbitae contained two exons (Fig. 2d).

Number of lncRNAs in tissues of female (a) and male (c) Zeugodacus cucurbitae. LncRNAs with null expression in female tissues (b) and male tissues (d) were discarded. Abbreviations were consistent with the above

Number of four types of lncRNA (a), the lncRNA length distribution (b), the isoform number of lncRNA (c), the exon number distribution of lncRNA (d). lincRNA means long intergenic non-coding RNA
Expression of lncRNAs in Z. cucurbitae
To analyze the differences in expression of lncRNAs among tissues, the hierarchical clustering of 1554 differentially expressed lncRNAs (DELs) was analyzed in a heatmap using the FPKM value (Fig. 3). Many DELs clustered in specific tissues based on lncRNA expression levels among the different tissues. DELs between every two pairs of tissues were analyzed. In female Z. cucurbitae. A total of 151 higher- and 103 lower-expressed lncRNAs were found in the comparison of Malpighian tubules vs. ovary. The comparison of midgut vs. fat body showed 69 DELs, among which 36 were higher- and 33 were lower-expressed (Fig. 4a). Comparisons of Malpighian tubules vs. testis and midgut vs. Malpighian tubules had the most and fewest DELs in males, respectively. A total of 806 DELs were found in male Malpighian tubules vs. testis; 604 were higher- and 202 were lower-expressed. A total of 45 DELs existed in midgut vs. Malpighian tubules of males; 28 were higher- and 17 were lower-expressed (Fig. 4b). DELs between similar tissues in male and female adults were analyzed. The comparison of ovary vs. testis had 623 DELs, which was much more than other tissue comparisons (Fig. 4c).

Cluster heatmap showing the expression profile of differentially expressed lncRNAs in female (a) and male (b) tissues of Zeugodacus cucurbitae. The heatmap was generated using R pheatmap. Red and Green indicate higher and lower expression levels, respectively. Abbreviations are consistent with the above

Statistical analysis of differentially expressed lncRNAs between tissues in female Zeugodacus cucurbitae (a), male Zeugodacus cucurbitae (b), and similar tissues between female and male Zeugodacus cucurbitae (c). Abbreviations are the same as above
LncRNAs showed differential expression among tissues. Tissue-specific lncRNAs were identified in all tissues. Venn diagrams showed that each tissue contained a certain number of tissue-specific lncRNAs. In midgut, Venn diagram analysis showed 8 and 8 specifically expressed lncRNAs in females and males (Fig. 5a1 and a2). A total of 5, 7, 9, and 21 specifically expressed lncRNAs were found in female Malpighian tubules (Fig. 5b1), male Malpighian tubules (Fig. 5b2), female fat body (Fig. 5c1), and male fat body (Fig. 5c2), respectively. A total of 42 ovary-specific lncRNAs had a relatively high expression in the ovary compared with other female tissues (Fig. 5d1). The number of testis-specific lncRNAs (364) was much larger than those of other tissues (Fig. 5d2).

Quantitative expression analysis of midgut, Malpighian tubules, fat body, ovary, and testis in Zeugodacus cucurbitae. Each section of the Venn diagrams shows the numbers of differentially expressed lncRNAs with a ratio of two tissues expression level above 10. Venn diagrams indicate the number of midgut-specific lncRNAs (a1 and a2), Malpighian tubules-specific lncRNAs (b1 and b2), fat body-specific lncRNAs (c1 and c2), ovary-specific lncRNAs (d1), and testis-specific lncRNAs (d2) in female and male Zeugodacus cucurbitae. Abbreviations are consistent with those used previously
Functional annotation of target genes of tissue-specific lncRNAs
GO and KEGG pathway analysis were conducted to study the potential functions of lncRNAs, and some of them can regulate the expression of neighboring genes (cis) and related co-expressed genes (trans) [31]. To illustrate some special functional annotations, target genes of tissue-specific (e.g., midgut-specific) lncRNAs were analyzed. A total of 457 target genes were obtained in the female midgut, among which 51 were cis-regulated and 410 were trans-regulated. For the male midgut, a total of 273 target genes were predicted, including 34 cis-regulated and 241 trans-regulated genes. GO analysis indicated that these target genes were involved in different physiological activities, including biological process, molecular function, and cellular component. In these categories, metabolic process, catalytic activity, and membrane were the most abundant subgroups (Fig. 6a). KEGG pathway analyses showed that these target genes were most frequently predicted in metabolism, among which the three pathways (purine metabolism, oxidative phosphorylation, and carbon metabolism) were most significantly enriched (Fig. 6b).

GO and KEGG pathway analyses of the target genes of midgut-specific lncRNAs in Zeugodacus cucurbitae. a GO analysis of the functions of lncRNA target genes. b KEGG pathway analysis of lncRNA target genes
Validation of differentially expressed lncRNAs
Four differentially expressed lncRNAs were randomly selected and their expression patterns in the eight tissues were examined by RT-qPCR. The selected four differentially expressed lncRNAs were named as Zc-Lnc22787, Zc-Lnc50977, Zc-Lnc99852, and Zc-Lnc11868. The expression patterns of these four lncRNAs calculated from RNA-seq data and RT-qPCR results were consistent (Fig. 7). All of our findings showed that our pipeline was strict in lncRNA identification and indicated that the identified lncRNAs were differentially expressed, in vivo.

Validation of four randomly selected differentially expressed lncRNAs by quantitative real-time PCR (RT-qPCR). The bar represents the mean lncRNA expression and the error bar represents the positive standard error (SE) of the mean. Abbreviations were consistent with those used previously. Data were analyzed by one-way ANOVA followed with Tukey’s test (P < 0.05)
Discussion
The lncRNAs are responsible for several key physiological processes [32, 33], including epigenetics [34], immune response [35], and protein degradation [36]. LncRNAs in insect species have now been studied in D. melanogaster [5], A. aegypti [24], B. mori [38]. After identification under a computational pipeline, the screening criteria of the expression threshold of at least 1 FPKM in each tissue resulted in a strict catalog containing 3124 lncRNAs. A similar result was reported in Drosophila, in which 1077 lncRNAs were identified from 43,967 transcripts in the transcriptomes of different development stages [5]. Each tissue had a specific number of lncRNA in Z. cucurbitae. In Drosophila, lncRNAs were also distributed in many tissues of males [18]. Differences in the lncRNA numbers of different tissues may explain the variable lncRNA amounts in different insect species. Among the identified lncRNAs, the long intergenic lncRNAs (lincRNA) were most common, followed by intronic, sense, and anti-sense lncRNAs. In B. mori, lincRNAs and intronic lncRNAs were the most and least common, and sense lncRNAs were not identified [50], the Coding-Potential Assessment Tool (CPAT version 1.2.2) [51], and Coding Potential Calculator (CPC, version 0.9 r2) [52] were used to predict the protein-coding potential. Transcripts with CNCI scores < 0, CPAT = “no”, and CPC scores < 0 were retained. After that, Pfam was implemented and transcripts that contained any known protein domains would be excluded [53]. Finally, the remaining transcripts were aligned with Rfam database, GtRNAdb database, Silva database, and Repbase database, respectively, to screen out other ncRNA, such as small nuclear RNA (snRNA), transfer RNA (tRNA), small nucleolar RNA (snoRNA) repeat sequences, and ribosomal RNA (rRNA) using Bowtie tools [54]. Genome map** rates revealed large differences among biological replicates of Malpighian tubules from female and male melon flies. Considering this, Malpighian tubules as well as other tissues had one deleted replicate, and the average FPKM values between the remaining two replicates were used for downstream analyses [20]. Transcripts with an FPKM value < 1 in all tissues were considered as null-expressed and were discarded. The remaining transcripts were considered reliable lncRNAs. Additionally, mRNAs were obtained from the same RNA-seq libraries in this study.
Tissue-specific expressed lncRNAs
Tissue-specific lncRNAs refer to lncRNAs that have extremely high expression in the given tissue [18]. To study the tissue-specific lncRNAs in female and male Z. cucurbitae, DESeq was used to analyze the significance of the differential expression of lncRNAs in each two tissues [55]. In this step, the software provided a statistical program for calculating the difference in numeric gene expression analysis with fold change ≥2 and a False Discovery Rate (FDR) < 0.05. On this basis, tissue-specific expressed lncRNAs were screened in each tissue with the ratio of FPKMtissue 1/FPKMall the others ≥ 10.
Target prediction and GO and KEGG pathway analysis
LncRNA targets were predicted according to the genomic location and co-expression between lncRNAs and mRNAs. Two categories (cis-regulation and trans-regulation) of the lncRNA regulation modes were analyzed. LncRNAs’ regulation on their neighbor genes within 100 kb upstream and downstream in chromosomes was regarded as cis-regulation [56]. For trans-regulation, co-expression analyses of lncRNA and mRNA were investigated based on their expressions as previously implemented in tissues of B. mori [26]. Coefficients with r > 0.9 or < − 0.9 and pearson’s correlation with p-value < 0.01 were judged to be correlated expressed. All of the identified cis- and trans-regulated protein-coding genes were used for GO and KEGG pathway analysis. TopGO R packages and KOBAS software [57] were used for GO and KEGG pathway analysis, respectively.
Quantitative real-time PCR (RT-qPCR)
To validate expression patterns of differentially expressed lncRNAs, the eight tissues were dissected from 5-day-old melon fly adults in the same manner as the sequenced samples. After total RNA isolation, lnRcute lncRNA cDNA kit (TIANGEN, Bei**g, China) was used for first-strand lncRNA cDNA synthesis. Primers used for lncRNAs validation were designed using Primer 3.0 (http://bioinfo.ut.ee/primer3-0.4.0/) (Tab. S1). To determine the cycle threshold (Ct) value and amplification efficiency of each pair of primers, a standard curve was conducted with serial dilutions of cDNA (1, 5− 1, 5− 2, 5− 3, 5− 4). The qPCR reaction was run on a CFX384 Optics Module (Bio-Rad, Singapore) using the lnRcute lncRNA SYBR Green premix (TIANGEN, Bei**g, China). RT-qPCR was conducted with 10 μL of mixture, each consisted of 5 μL of lncRNA SYBR premix, 4 μL of nuclease-free water, 0.5 μL of lncRNA cDNA (~ 500 ng/μL), and 0.25 μL each of forward and reverse primers (10 μM). The PCR procedure was as follows: an initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 15 s, the specificity of primers were ensured by the record of a melting curve analysis from 60 °C to 95 °C. Relative expression levels of lncRNAs among different tissues were normalized by Alpha-tubulin and beta-tubulin 1 [58]. All experiments were conducted in four biological replicates. Data were calculated by qBase plus software [59].
Statistical analysis
The difference among tissues was analyzed using SPSS 19.0 software (IBM, Chicago, IL, USA) with a one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference (HSD) test (P < 0.05).
Availability of data and materials
All of the RNA sequencing data produced in the current study are available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under the accession number: PRJNA579200.
Abbreviations
- DEL:
-
differentially expressed lncRNA
- FB:
-
fat body
- FDR:
-
false discovery rate
- FPKM:
-
fragments per kilobase per million reads
- LincRNA:
-
long intergenic non-coding RNA
- LncRNA:
-
long non-coding RNA
- MG:
-
midgut
- MT:
-
Malpighian tubules
- nt:
-
nucleotides
- OV:
-
ovary
- TE:
-
testis
- vs.:
-
versus
References
**ao H, Yuan Z, Guo D, Hou B, Yin C, Zhang W, Li F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genomics. 2015;16:749.
Kodama Y, Shumway M, Leinonen R. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40(1):54–6.
Yang L, Froberg JE, Lee JT. Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem Sci. 2014;39(1):35–43.
Li BJ, Jiang DL, Meng ZN, Zhang Y, Zhu ZX, Lin HR, **a JH. Genome-wide identification and differentially expression analysis of lncRNAs in tilapia. BMC Genomics. 2018;19(1):729.
Chen B, Zhang Y, Zhang X, Jia SL, Chen S, Kang L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci Rep. 2016;6:23330.
Zhu B, Xu MY, Shi HY, Gao XW, Liang P. Genome-wide identification of lncRNAs associated with chlorantraniliprole resistance in diamondback moth Plutella xylostella (L.). BMC Genomics. 2017;18(1):380.
Liu FL, Guo DH, Yuan ZT, Chen C, **ao HM. Genome-wide identification of long non-coding RNA genes and their association with insecticide resistance and metamorphosis in diamondback moth, Plutella xylostella. Sci Rep. 2017;7(1):1–11.
Laurent GS, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet. 2015;31(5):239–51.
Chen DF, Chen HZ, Du Y, Zhou DD, Geng SH, Wang HP, Wan JQ, **ong CL, Zheng YZ, Guo R. Genome-wide identification of long non-coding RNAs and their regulatory networks involved in Apis mellifera ligustica response to Nosema ceranae infection. Insects. 2019;10(8):245.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–89.
Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10(6):924–33.
Zhan S, Dong Y, Zhao W, Guo J, Zhong T, Wang L, Li L, Zhang H. Genome-wide identification and characterization of long non-coding RNAs in developmental skeletal muscle of fetal goat. BMC Genomics. 2016;17:666.
Gopalakrishnan K, Kumarasamy S, Mell B, Joe B. Genome-wide identification of long noncoding RNAs in rat models of cardiovascular and renal disease. Hypertens. 2015;65(1):200–10.
Kaushik K, Leonard VE, Shamsudheen K, Lalwani MK, Jalali S, Patowary A, Joshi A, Scaria V, Sivasubbu S. Dynamic expression of long non-coding RNAs (lncRNAs) in adult zebrafish. PLoS One. 2013;8(12):e83616.
Arriaga CC, Fonseca GY, Valdes QC, Arzate MR, Guerrero G, Recillas TF. A long non-coding RNA promotes full activation of adult gene expression in the chicken α-globin domain. Epigenetics. 2014;9(1):173–81.
Wang ZX, Jiang YY, Wu H, **e XY, Huang B. Genome-wide identification and functional prediction of long noncoding RNAs involved in the heat stress response in Metarhizium robertsii. Front Microbiol. 2019;10:2336.
Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512(7515):393–9.
Wang Y, Xu T, He W, Shen X, Zhao Q, Bai J, You M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics. 2018;110(1):35–42.
Jenkins AM, Waterhouse RM, Muskavitch MA. Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genomics. 2015;16(1):337.
Li MZ, **ao HM, He K, Li F. Progress and prospects of noncoding RNAs in insects. J Integr Agric. 2019;18(4):729–47.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon VB, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–27.
Etebari K, Asad S, Zhang GM, Asgari S. Identification of Aedes aegypti long intergenic non-coding RNAs and their association with Wolbachia and dengue virus infection. PLoS Neglected Trop Dis. 2016;10(10):e0005069.
Zhang X, Xu YN, Chen B, Kang L. Long noncoding RNA PAHAL modulates locust behavioural plasticity through the feedback regulation of dopamine biosynthesis. PLoS Genet. 2020;16(4):29.
Wu Y, Cheng T, Liu C, Liu D, Zhang Q, Long R, Zhao P, **a Q. Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori. PLoS One. 2016;11(1):e0147147.
Koyama J, Kakinohana H, Miyatake T. Eradication of the melon fly, Bactrocera cucurbitae, in Japan: importance of behavior, ecology, genetics, and evolution. Annu Rev Entomol. 2004;49:331–49.
Khan MH, Khuhro NH, Awais M, Memon RM, Asif MU. Functional response of the pupal parasitoid, Dirhinus giffardii towards two fruit fly species, Bactrocera zonata and B. cucurbitae. Entomol Gen. 2020;40(1):87–95.
Sim SB, Geib SM. A chromosome-scale assembly of the Bactrocera cucurbitae genome provides insight to the genetic basis of white pupae. G3: Genes Genomes Genet. 2017;7(6):1927–40.
Wei D, Xu HQ, Chen D, Zhang SY, Li WJ, Smagghe G, Wang JJ. Genome-wide gene expression profiling of the melon fly, Zeugodacus cucurbitae, during thirteen life stages. Sci Data. 2020;7(1):45.
Huang MG, Zhong ZY, Lv MX, Shu J, Tian Q, Chen JX. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget. 2016;7(30):47186.
Satyavathi V, Ghosh R, Subramanian S. Long non-coding RNAs regulating immunity in insects. Non-coding RNA. 2017;3(1):14.
Chang ZX, Ajayi OE, Guo DY, Wu QF. Genome-wide characterization and developmental expression profiling of long non-coding RNAs in Sogatella furcifera. Insect Sci. 2019;00:1–11.
Hassan MQ, Tye CE, Stein GS, Lian JB. Non-coding RNAs: epigenetic regulators of bone development and homeostasis. Bone. 2015;81:746–56.
Peng XX, Gralinski L, Armour CD, Ferris MT, Thomas MJ, Proll S, Bradel TBG, Korth MJ, Castle JC, Biery MC. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio. 2010;1(5):00206–10.
Humann FC, Tiberio GJ, Hartfelder K. Sequence and expression characteristics of long noncoding RNAs in honey bee caste development–potential novel regulators for transgressive ovary size. PLoS One. 2013;8(10):e78915.
Petrella V, Aceto S, Musacchia F, Colonna V, Robinson M, Benes V, Cicotti G, Bongiorno G, Gradoni L, Volf P, et al. De novo assembly and sex-specific transcriptome profiling in the sand fly Phlebotomus perniciosus (Diptera, Phlebotominae), a major old world vector of Leishmania infantum. BMC Genomics. 2015;16(1):847.
Bryant WB, Ray S, Mills MK. Global analysis of small non-coding RNA populations across tissues in the malaria vector, Anopheles gambiae. Insects. 2020;11(7):406.
Chen MJM, Chen LK, Lai YS, Lin YY, Wu DC, Tung YA, Liu KY, Shih HT, Chen YJ, Lin YL. Integrating RNA-seq and ChIP-seq data to characterize long non-coding RNAs in Drosophila melanogaster. BMC Genomics. 2016;17(1):220.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22(9):1760–74.
Wen K, Yang L, **ong T, Di C, Ma D, Wu M, Xue Z, Zhang X, Long L, Zhang W, et al. Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res. 2016;26(9):1233–44.
Kern C, Wang Y, Chitwood J, Korf I, Delany M, Cheng H, Medrano JF, Van Eenennaam AL, Ernst C, Ross P. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genomics. 2018;19(1):684.
Li MX, Wen SY, Guo XQ, Bai BY, Gong ZF, Liu XJ, Wang YJ, Zhou YQ, Chen XW, Liu L. The novel long non-coding RNA CRG regulates Drosophila locomotor behavior. Nucleic Acids Res. 2012;40(22):11714–27.
Padron A, Molina CA, Quinones M, Ribeiro JMC, Ramphul U, Rodrigues J, Shen K, Haile A, Ramirez JL, Barillas MC. In depth annotation of the Anopheles gambiae mosquito midgut transcriptome. BMC Genomics. 2014;15:636.
Yu HZ, Li NY, Zeng XD, Song JC, Yu XD, Su HN, Chen CX, Yi L, Lu ZJ. Transcriptome analyses of Diaphorina citri midgut responses to candidatus liberibacter asiaticus infection. Insects. 2020;11(3):171.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223.
Zheng Y, Bi J, Hou MY, Shen W, Zhang W, Ai H, Yu XQ, Wang YF. Ocnus is essential for male germ cell development in Drosophila melanogaster. Insect Mol Biol. 2018;27(5):545–55.
Sun L, Goff LA, Trapnell C, Alexander R, Lo KA, Hacisuleyman E, Sauvageau M, Tazon VB, Kelley DR, Hendrickson DG. Long noncoding RNAs regulate adipogenesis. Proc Natl Acad Sci. 2013;110(9):3387–92.
Wang LG, Park HJ, Dasari S, Wang SQ, Kocher JP, Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74.
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei LP, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(2):345–9.
Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43(1):30–8.
Langmead B. Aligning short sequencing reads with bowtie. Curr Protoc Bioinf. 2010;32(1):711–4.
Anders S. Analysing RNA-seq data with the DESeq package. Mol Biol. 2010;43(4):1–17.
Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the develo** brain. PLoS Genet. 2009;5(8):e1000617.
**e C, Mao XZ, Huang JJ, Ding Y, Wu JM, Dong S, Kong L, Gao G, Li CY, Wei LP. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:316–22.
Li WJ, Song YJ, Xu HQ, Wei D, Wang JJ. Vitelline membrane protein gene ZcVMP26Ab and its role in preventing water loss in Zeugodacus cucurbitae (Coquillett) embryos. Entomol Gen. 2020;in press.
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8(2):R19.
Acknowledgments
We thank Dr. Fei Pan from Institute of Plant Protection, Hainan Academy of Agricultural Sciences, Hainan, China, for his kind assistant with collecting the insects.
Funding
This study was supported by the Foundation Project of Southwest University (SWU019033), the Fundamental Research Funds for the Central Universities (XDJK2020C081), and the earmarked fund for Modern Agro-industry (Citrus) Technology Research System of China (CARS-26).
Author information
Authors and Affiliations
Contributions
D.W., G.S. and J.J.W. conceived and designed the project; W.J.L., Y.J.S. and H.L.H. contributed to the sample collection, tissue dissection, and RNA isolation; W.J.L., Y.J.S. and H.Q.X. contributed to the data analysis. W.J.L. and D.W. wrote the manuscript; G.S. and J.J.W. revised the writing. All of the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Zeugodacus cucurbitae is one of the most common agricultural pests, which is not included in the “List of Endangered and Protected Animals in China”. The original collecting site is not private and the field collection of Zeugodacus cucurbitae was permitted.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
The computational pipeline for lncRNA identification from transcriptome.
Additional file 2: Table S1.
Primer sequences used for RT-qPCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, WJ., Song, YJ., Han, HL. et al. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Genomics 21, 600 (2020). https://doi.org/10.1186/s12864-020-07014-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07014-x