
Abstract
Background
Anti-CRISPR proteins are potent modulators that inhibit the CRISPR-Cas immunity system and have huge potential in gene editing and gene therapy as a genome-editing tool. Extensive studies have shown that anti-CRISPR proteins are essential for modifying endogenous genes, promoting the RNA-guided binding and cleavage of DNA or RNA substrates. In recent years, identifying and characterizing anti-CRISPR proteins has become a hot and significant research topic in bioinformatics. However, as most anti-CRISPR proteins fall short in sharing similarities to those currently known, traditional screening methods are time-consuming and inefficient. Machine learning methods could fill this gap with powerful predictive capability and provide a new perspective for anti-CRISPR protein identification.
Results
Here, we present a novel machine learning ensemble predictor, called PreAcrs, to identify anti-CRISPR proteins from protein sequences directly. Three features and eight different machine learning algorithms were used to train PreAcrs. PreAcrs outperformed other existing methods and significantly improved the prediction accuracy for identifying anti-CRISPR proteins.
Conclusions
In summary, the PreAcrs predictor achieved a competitive performance for predicting new anti-CRISPR proteins in terms of accuracy and robustness. We anticipate PreAcrs will be a valuable tool for researchers to speed up the research process. The source code is available at: https://github.com/Lyn-666/anti_CRISPR.git.
Similar content being viewed by others
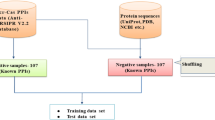


Background
CRISPR-Cas adaptive immune system is one of the most widespread immunity strategies in prokaryotes against invading bacteriophages and plasmids [1, 2]. To counteract and overcome different CRISPR-Cas immunity systems, bacteriophages have evolved anti-CRISPR proteins (Acrs) that were first discovered in Pseudomonas aeruginosa phages in 2013 [3]. Subsequently, a proliferation of Acrs has proved to inactivate multiple CRISPR subtypes [3,4,5,6,7].
Several methods have been proposed to identify Acrs, including “Guilt-by-association” studies [6, 8], self-targeting CRISPR arrays [6, 7], and metagenome DNA screening [9, 10], etc. These methods assumed the new Acrs are similar to the previous Acrs. However, most Acrs fall short in sharing similarities currently acknowledged. Therefore, the traditional screening methods based on homology search are unreliable and require a lot of prior knowledge of Acrs to identify new Acrs. For instance, the “Guilt-by-association” method involves searching for homologs of helix-turn-helix (HTH)-containing proteins that are typically encoded downstream of Acrs [11]. The performance of “Guilt-by-association” is unstable when known Acrs proteins might share low similarity with queried protein. Therefore, a computational approach with less requirement for prior knowledge of known Acrs will provide a new perspective on the identification of Acrs. Machine learning algorithms with appropriate features could reveal the potential mechanism of Acrs and identify the Acrs without prior knowledge.
Recently, some machine learning methods have been presented for predicting Acrs. There are several web servers about Acrs, such as: Anti-CRISPRdb [12], AcrHub [13], AcrDB [14], CRISPRminer2 [15], AcRanker [14, 16], AcrFinder [17], AcrCatalog [18] and PaCRISPR [48], the original PSSM profile (L × 20) could be reduced to a L × 10 matrix by merging some columns. RPSSM is obtained by exploring the local sequence information based on the L × 10 reduced PSSM [49, 50]:
and
where \(p_{A} ,p_{R} , \ldots ,p_{V}\) represent the 20 columns in the original PSSM profile corresponding to the 20 amino acids. The re-PSSM is further transformed into a 10-dimensional vector:
and
Additionally, the re-PSSM can be further transformed into a 10 × 10 matrix to capture the local sequence-order information by this formula:
where \({p}_{i,j}\) represents the element at the ith row and jth column of there-PSSM. Finally, a 110-dimensional RPSSM feature is obtained by combining \({E}_{j,t}\) and \({E}_{j}\):
Pretrained SSA embedding
The pretrained SSA embedding mosdel is obtained by combining the pre-trained language model with the soft sequence alignment (SSA) [51]. First, an embedding matrix RL×121 is given using the stacked BiLSTM encoders for each sequence, where L is the protein sequence length [52]. Then, the pretrained SSA embedding model is trained and optimized by SSA, which the following formulas could describe. For convenience, we supposed two embedding matrices P1(RL1×121) and P2(RL2×121), of two different protein sequences with lengths L1 and L2, respectively:
where xi, yi are vectors with 121-dimension.
The following formula represents the similarity of P1 and P2:
and
with
The SSA embedding model could convert each protein sequence into an embedded matrix RL×121, and finally, an average pooling operation obtained a 121-dimensional feature.
Feature selection
Original features are represented by a high dimensional vector or matrix, which would raise severe problems in machine learning algorithms, such as overfitting, time-consuming training process and high requirement of computing resources. Therefore, identifying the most contributing information and features plays a vital role in performance improvement. As one of the most popular feature selection algorithms, maximum relevance minimum redundancy (mRMR) was proposed by Peng et al. [53] and has been applied in many studies and achieved robust performances [54,55,56]. In this study, mRMR was used to identify the most important features and improve the generalization ability of the model.
Machine learning algorithm
In this study, we focused on the traditional machine learning classification methods, including support vector machine, k-nearest neighbor, multi-layer perceptron, logistic regression, random forest, extreme gradient boosting, Light gradient boost machine and ensemble method that integrates the previous eight classification methods by hard voting strategy and stacking classifiers. More information is shown in the following subsections.
Support vector machine
Support vector machine (SVM) was first proposed by Vapnik et al. [57], and has successfully dealt with some binary classification problems in bioinformatics [25, 58, 59]. Two parameters Cost (C) and Gamma (γ) affect the performance of the SVM model with the RBF kernel. In this study, we used the grid search strategy to optimize C and γ in the space {2−6, 2−5, …, 25, 26}. Finally, an SVM classifier with the optimal value of C and γ was constructed.
K-nearest neighbor
K-nearest neighbor (KNN) is a fundamental classifier that has been applied in predicting protein function [60], extracting protein–protein information [61], and predicting eukaryotic protein subcellular [62]. The performance of KNN is directly affected by the parameter k. In this study, a grid search within the space \(\left\{ {1,2, \ldots ,\max \left\{ {\sqrt {FeaNum} ,\frac{FeaNum}{2}} \right\}} \right\}\) was applied to optimize the parameter k during model training, where FeaNum is the number of features used in modelling.
Multi-layer perception
Multi-layer perceptron (MLP) is known as a type of artificial neural network (ANN) [63, 64]. MLP has been applied in many bioinformatics studies, such as the prediction of protein structure classes [65], protein tertiary structure [66], and DNA–protein binding sites [67]. In this study, an MLP classifier with two hidden layers was trained, and the first and second hidden layers have 64 and 32 nodes, respectively. The maximum learning iterations is 1000.
Logistic regression
Logistic regression (LR) is widely used to predict the probability of an event happening [59, 68], which the following formula could represent:
where p(y) is the expected probability of dependent variable \(\mathrm{y}\), and β0 and β1 are constants.
Random forest
Random forest (RF) classifier is proposed by Breiman [69] and has been used in the prediction of type IV secreted effector proteins [70] and protein structural class [59]. To find the optimal number of the trees M and features mtry, we used a gird searching to optimize \(\mathrm{M}\) and \(\mathrm{mtry}\) within space \(\{1, 2,\cdots ,\mathrm{max}\left\{\sqrt{FeaNum},\frac{FeaNum}{2}\right\}\}\) and {1, 6, 11, 16}, respectively, where FeaNum is the number of features adopted during modeling.
XGBoost
Extreme gradient boosting (XGBoost) is a scalable end-to-end tree boosting system [71] and has been widely used as a fast and highly effective machine learning method [72, 73]. Eitzinger et al. implemented AcRanker using XGBoost to identify Acrs [14, 16]. In this study, the default parameters are adopted in the XGBoost model, except for the learning rate of 0.1.
LightGBM
Light gradient boost machine (LightGBM) shows excellent performance when the feature dimension is high and the larger data size [21]. LightGBM has been used in identifying miRNA targets [74] and predicting the protein–protein interactions [75] and the blood–brain-barrier penetration [76]. This study used the LightGBM package with default parameters in python during experiments.
CatBoost
CatBoost achieves state-of-the-art results since it successfully handles categorical features and calculates leaf values via a new scheme, which helps reduce overfitting [23]. Catboost has been applied in various tasks, including molecular structure relationship and the biological activity prediction [77] and the identification of pyroptosis-related molecular subtypes of lung adenocarcinoma [78]. In this study, the parameters of CatBoost were set as default values.
Ensemble learning method
This study proposed three ensemble models to construct more robust and reliable classifiers, which predicted new Acrs proteins by integrating the above eight classifiers (SVM, KNN, MLP, LR, RF, XGB, LightGBM, and CatBoost) through the hard voting rule (Ens-vote) or two stacking classifiers with logistic regression (Sta-LR) and gradient boosting classifier (Sta-GBC) [79], respectively.
Performance assessment
Fairly evaluating the classification methods' predictive performance is an essential subject in machine learning. In this study, we used six measurements, namely, Sensitivity (SN), Specificity (SP), Accuracy (ACC), Precision (PRE), F1-score, and Matthew’s correlation coefficient (MCC) [80], which are denoted as:
where TP, TN, FP, and FN are the number of true positive, true negative, false positive and false negative, respectively. Besides, the area under the receiver operating characteristic (ROC) curve (AUC) is also used to assess the performance, and the ROC was shown in a plot of the TP rate versus the FP rate. All methods were evaluated based on a fivefold cross-validation.
Availability of data and materials
The datasets of this study are available on Github (https://github.com/Lyn-666/anti_CRISPR.git).
Abbreviations
- Acrs:
-
Anti-CRISPR proteins
- AAC:
-
Amino acid composition
- PAAC:
-
Pseudo-amino acid composition
- PSSM:
-
Position specific scoring matrix
- PSSM-AC:
-
Position-specific matrix auto covariance
- RPSSM:
-
Reduced position specific scoring matrix
- SSA:
-
Soft sequence alignment
- mRMR:
-
Maximum relevance minimum redundancy
- SVM:
-
Support vector machine
- KNN:
-
K-nearest neighbor
- MLP:
-
Multi-layer perceptron
- LR:
-
Logistic regression
- RF:
-
Random forest
- XGBoost:
-
Extreme gradient boosting
- LightGBM:
-
Light gradient boost machine
- SN:
-
Sensitivity
- SP:
-
Specificity
- PRE:
-
Precision
- TP:
-
True positive
- FP:
-
False positive
- TN:
-
True negative
- FN:
-
False negative
- MCC:
-
Matthews correlation coefficient
- AUC:
-
Area under the ROC curve
- ACC:
-
Accuracy
- PRC:
-
Precision-recall curve
- ROC:
-
Receiver operating characteristic
- AUPRC:
-
Area under the PRC
References
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12.
Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322(5909):1843–5.
Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493(7432):429–32.
Pawluk A, Davidson AR, Maxwell KL. Anti-CRISPR: discovery, mechanism and function. Nat Rev Microbiol. 2018;16(1):12–7.
Stanley SY, Maxwell KL. Phage-encoded anti-CRISPR defenses. Annu Rev Genet. 2018;52:445–64.
Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science. 2018;362(6411):240–2.
Watters KE, Fellmann C, Bai HB, Ren SM, Doudna JA. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science. 2018;362(6411):236–9.
Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, Fineran PC, Maxwell KL, Davidson AR. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016;1(8):1–6.
Uribe RV, Van Der Helm E, Misiakou M-A, Lee S-W, Kol S, Sommer MOA. Discovery and characterization of Cas9 inhibitors disseminated across seven bacterial phyla. Cell Host Microbe. 2019;25(2):233-241.e235.
Forsberg KJ, Bhatt IV, Schmidtke DT, Javanmardi K, Dillard KE, Stoddard BL, Finkelstein IJ, Kaiser BK, Malik HS. Functional metagenomics-guided discovery of potent Cas9 inhibitors in the human microbiome. Elife. 2019. https://doi.org/10.7554/eLife.46540.
Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. Naturally occurring off-switches for CRISPR-Cas9. Cell. 2016;167(7):1829–38.
Dong C, Hao G-F, Hua H-L, Liu S, Labena AA, Chai G, Huang J, Rao N, Guo F-B. Anti-CRISPRdb: a comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 2018;46(D1):D393–8.
Wang J, Dai W, Li J, Li Q, ** anti-CRISPR proteins. Nucleic Acids Res. 2020;49(D1):D630–8.
Huang L, Yang B, Yi H, Asif A, Wang J, Lithgow T, Zhang H, Minhas A, Ul Amir F, Yanbin Y. AcrDB: a database of anti-CRISPR operons in prokaryotes and viruses. Nucleic Acids Re. 2021;49(D1):D622–9.
Zhang F, Zhao S, Ren C, Zhu Y, Zhou H, Lai Y, Zhou F, Jia Y, Zheng K, Huang Z. CRISPRminer is a knowledge base for exploring CRISPR-Cas systems in microbe and phage interactions. Commun Biol. 2018. https://doi.org/10.1038/s42003-018-0184-6.
Eitzinger S, Asif A, Watters KE, Iavarone AT, Knott GJ, Doudna JA, Minhas A, Ul Amir F. Machine learning predicts new anti-CRISPR proteins. Nucleic Acids Res. 2020;48(9):4698–708.
Yi H, Huang L, Yang B, Gomez J, Zhang H, Yin Y. AcrFinder: genome mining anti-CRISPR operons in prokaryotes and their viruses. Nucleic Acids Res. 2020;48(W1):W358–65.
Gussow AB, Shmakov SA, Makarova KS, Wolf YI, Bondy-Denomy J, Koonin EV. Vast diversity of anti-CRISPR proteins predicted with a machine-learning approach. Spring Harbor: Cold Spring Harbor Laboratory; 2020.
Wang J, Dai W, Li J, **e R, Dunstan RA, Stubenrauch C, Zhang Y, Lithgow T. PaCRISPR: a server for predicting and visualizing anti-CRISPR proteins. Nucleic Acids Res. 2020;48(W1):W348–57.
Gussow AB, Park AE, Borges AL, Shmakov SA, Makarova KS, Wolf YI, Bondy-Denomy J, Koonin EV. Machine-learning approach expands the repertoire of anti-CRISPR protein families. Nat Commun. 2020. https://doi.org/10.1038/s41467-020-17652-0.
Fernández-Delgado M, Cernadas E, Barro S, Amorim D. Do we need hundreds of classifiers to solve real world classification problems? J Mach Learn Res. 2014;15(1):3133–81.
Ke G, Meng Q, Finley T, Wang T, Chen W, Ma W, Ye Q, Liu T-Y. Lightgbm: a highly efficient gradient boosting decision tree. In: Advances in neural information processing systems. 2017, p. 30.
Dorogush AV, Ershov V, Gulin A. CatBoost: gradient boosting with categorical features support 2018. ar**v preprint https://arxiv.org/abs/1810.11363.
Zou L, Chen K. Computational prediction of bacterial type IV-B effectors using C-terminal signals and machine learning algorithms. In: 2016 IEEE conference on computational intelligence in bioinformatics and computational biology (CIBCB). IEEE;2016.
Zou L, Nan C, Hu F. Accurate prediction of bacterial type IV secreted effectors using amino acid composition and PSSM profiles. Bioinformatics. 2013;29(24):3135–42.
Wang Y, Wei X, Bao H, Liu S-L. Prediction of bacterial type IV secreted effectors by C-terminal features. BMC Genom. 2014;15(1):50.
Chen Z, Zhou Y, Song J, Zhang Z. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim Biophys Acta BBA Proteins Proteom. 2013;1834(8):1461–7.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10(1):421.
Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–2.
Isik Z, Yanikoglu B, Sezerman U. Protein structural class determination using support vector machines. In: Aykanat C, Dayar T, Körpeoğlu İ, editors. Computer and information sciences—ISCIS 2004. Berlin, Heidelberg: Springer; 2004. p. 82–9. https://doi.org/10.1007/978-3-540-30182-0_9.
Chou K-C. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteom. 2009;6(4):262–74.
Bernardes J. A review of protein function prediction under machine learning perspective. Recent Patents Biotechnol. 2013;7(2):122–41.
Li F, Li C, Marquez-Lago TT, Leier A, Akutsu T, Purcell AW, Ian Smith A, Lithgow T, Daly RJ, Song J, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics. 2018;34(24):4223–31.
Li F, Chen J, Leier A, Marquez-Lago T, Liu Q, Wang Y, Revote J, Smith AI, Akutsu T, Webb GI, et al. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics. 2020;36(4):1057–65.
Li F, Leier A, Liu Q, Wang Y, **ang D, Akutsu T, Webb GI, Smith AI, Marquez-Lago T, Li J. Procleave: predicting protease-specific substrate cleavage sites by combining sequence and structural information. Genom Proteom Bioinform. 2020;18(1):52–64.
Mei S, Li F, **ang D, Ayala R, Faridi P, Webb GI, Illing PT, Rossjohn J, Akutsu T, Croft NP, et al. Anthem: a user customised tool for fast and accurate prediction of binding between peptides and HLA class I molecules. Brief Bioinform. 2021;22(5):bbaa415.
Wang X, Li F, Xu J, Rong J, Webb GI, Ge Z, Li J, Song J. ASPIRER: a new computational approach for identifying non-classical secreted proteins based on deep learning. Brief Bioinform. 2022;23(2):bbac031.
Li F, Guo X, **ang D, Pitt ME, Bainomugisa A, Coin LJ. Computational analysis and prediction of PE_PGRS proteins using machine learning. Comput Struct Biotechnol J. 2022;20:662–74.
Wang X-F, Gao P, Liu Y-F, Li H-F, Lu F. Predicting thermophilic proteins by machine learning. Curr Bioinform. 2020;15(5):493–502.
Chen H, Li F, Wang L, ** Y, Chi C-H, Kurgan L, Song J, Shen J. Systematic evaluation of machine learning methods for identifying human–pathogen protein–protein interactions. Brief Bioinform. 2021;22(3):bbaa068.
Chou K-C, Zhang C-T. Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995;30(4):275–349.
Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct Funct Bioinform. 2001;43(3):246–55.
Chou K-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21(1):10–9.
Chen Z, Zhao P, Li C, Li F, **ang D, Chen Y-Z, Akutsu T, Daly J, Roger WI, Geoffrey ZQ, et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021;49(10):e60–e60.
Wold S, Jonsson J, Sjörström M, Sandberg M, Rännar S. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal Chim Acta. 1993;277(2):239–53.
Liu T, Zheng X, Wang C, Wang J. Prediction of subcellular location of apoptosis proteins using pseudo amino acid composition: an approach from auto covariance transformation. Protein Pept Lett. 2010;17(10):1263–9.
Wang J, Yang B, Revote J, Leier A, Marquez-Lago TT, Webb G, Song J, Chou K-C, Lithgow T. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics. 2017;33(17):2756–8.
Li T, Fan K, Wang J, Wang W. Reduction of protein sequence complexity by residue grou**. Protein Eng Des Sel. 2003;16(5):323–30.
Ding S, Li Y, Shi Z, Yan S. A protein structural classes prediction method based on predicted secondary structure and PSI-BLAST profile. Biochimie. 2014;97:60–5.
Ding C, Han H, Li Q, Yang X, Liu T. iT3SE-PX: identification of bacterial type III secreted effectors using PSSM profiles and XGBoost feature selection. Comput Math Methods Med. 2021. https://doi.org/10.1155/2021/6690299.
Bepler T, Berger B. Learning protein sequence embeddings using information from structure. 2019. https://arxiv.org/abs/1902.08661.
Lv Z, Cui F, Zou Q, Zhang L, Xu L. Anticancer peptides prediction with deep representation learning features. Brief Bioinform. 2021. https://doi.org/10.1093/bib/bbab008.
Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27(8):1226–38.
Li W, Lin K, Feng K, Cai Y. Prediction of protein structural classes using hybrid properties. Mol Divers. 2008;12(3–4):171–9.
Ni Q, Chen L. A feature and algorithm selection method for improving the prediction of protein structural class. Comb Chem High Throughput Screen. 2017;20(7):612–21.
Xu Y, Ding Y-X, Ding J, Wu L-Y, Xue Y. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Sci Rep. 2016;6(1):38318.
Boser BE, Guyon IM, Vapnik VN. A training algorithm for optimal margin classifiers. In: Proceedings of the fifth annual workshop on Computational learning theory—COLT '92. ACM Press; 1992.
Yang ZR. Biological applications of support vector machines. Brief Bioinform. 2004;5(4):328–38.
Wang J, Yang B, An Y, Marquez-Lago T, Leier A, Wilksch J, Hong Q, Zhang Y, Hayashida M, Akutsu T, et al. Systematic analysis and prediction of type IV secreted effector proteins by machine learning approaches. Brief Bioinform. 2019;20(3):931–51.
Lan L, Djuric N, Guo Y, Vucetic S. MS-k NN: protein function prediction by integrating multiple data sources. BMC Bioinform. 2013;14(S3):1–10.
Li L, **g L, Huang D. Protein-protein interaction extraction from biomedical literatures based on modified SVM-KNN. In: 2009 International conference on natural language processing and knowledge engineering. IEEE;2009.
Chou K-C, Shen H-B. Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-nearest neighbor classifiers. J Proteome Res. 2006;5(8):1888–97.
Bishop CM. Neural networks for pattern recognition. Oxford: Oxford University Press; 1995.
Tu JV. Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes. J Clin Epidemiol. 1996;49(11):1225–31.
Bao W, Chen Y, Wang D. Prediction of protein structure classes with flexible neural tree. Bio-med Mater Eng. 2014;24(6):3797–806.
Shao G, Chen Y. Predict the tertiary structure of protein with flexible neural tree. In: Huang D-S, Ma J, Kang-Hyun Jo M, Gromiha M, editors. Intelligent Computing Theories and Applications. Berlin, Heidelberg: Springer; 2012. p. 324–31.
Zeng H, Edwards MD, Liu G, Gifford DK. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics. 2016;32(12):i121–7.
LaValley MP. Logistic regression. Circulation. 2008;117(18):2395–9.
Breiman L. Random Forests. Mach Learning. 2001;45(1):5–32.
Wei L, Liao M, Gao X, Zou Q. An improved protein structural classes prediction method by incorporating both sequence and structure information. IEEE Trans NanoBiosci. 2015;14(4):339–49.
Chen T, Guestrin C. XGBoost. In: Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. ACM; 2016.
Li W, Yin Y, Quan X, Zhang H. Gene expression value prediction based on XGBoost algorithm. Front Genet. 2019;10:1077.
Zhong J, Sun Y, Peng W, **e M, Yang J, Tang X. XGBFEMF: an XGBoost-based framework for essential protein prediction. IEEE Trans NanoBiosci. 2018;17(3):243–50.
Wang D, Zhang Y, Zhao Y. LightGBM: an effective miRNA classification method in breast cancer patients. In: Proceedings of the 2017 international conference on computational biology and bioinformatics. 2017, p. 7–11.
Chen C, Zhang Q, Ma Q, Yu B. LightGBM-PPI: predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom Intell Lab Syst. 2019;191:54–64.
Shaker B, Yu M-S, Song JS, Ahn S, Ryu JY, Oh K-S, Na D. LightBBB: computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics. 2021;37(8):1135–9.
Hamzah H, Bustamam A, Yanuar A, Sarwinda D. Predicting the molecular structure relationship and the biological activity of dpp-4 inhibitor using deep neural network with Catboost method as feature selection. In: 2020 International conference on advanced computer science and information systems (ICACSIS). IEEE; 2020, pp. 101–108.
** LL, Lu L, Zhao Q, Kou Q, Wu X, Jiang Z, Rong G, Luo Y, Zhao Q. Identification and validation of the pyroptosis-related molecular subtypes of lung adenocarcinoma by bioinformatics and machine learning. Front Cell Dev Biol. 2021. https://doi.org/10.3389/fcell.2021.756340.
Alexandropoulos SAN, Aridas CK, Kotsiantis SB, Vrahatis MN. Stacking strong ensembles of classifiers. In: IFIP International Conference on Artificial Intelligence Applications and Innovations. Springer, Cham. 2019; pp. 545–556.
Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta BBA Protein Struct. 1975;405(2):442–51.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Australian Research Council (ARC) (LP110200333 and DP120104460), National Health and Medical Research Council of Australia (NHMRC) (1092262, 490989), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), and a Major Inter-Disciplinary Research (IDR) Grant Awarded by Monash University. C.L. is currently supported by an NHMRC CJ Martin Early Career Research Fellowship (1143366).
Author information
Authors and Affiliations
Contributions
LZ and XW conceived the project and designed the experiments. LZ performed the model construction, data analysis and drafted the manuscript. FL and JS provided useful comments and assisted with the data analysis and model construction. All authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
J.S. is an Associate Editor of BMC Bioinformatics. LZ, XW and FL declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Performance of all single features.
Additional file 1: Table S2.
Performance of ensemble features.
Additional file 1: Table S3.
Performance of combinational features.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, L., Wang, X., Li, F. et al. PreAcrs: a machine learning framework for identifying anti-CRISPR proteins. BMC Bioinformatics 23, 444 (2022). https://doi.org/10.1186/s12859-022-04986-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-022-04986-3