
Abstract
Introduction
The coexistence of different molecular types of classical protease-resistant prion protein in the same individual have been described, however, the simultaneous finding of these with the recently described protease-sensitive variant or variably protease-sensitive prionopathy has, to the best of our knowledge, not yet been reported.
Case presentation
A 74-year-old Caucasian woman showed a sporadic Creutzfeldt–Jakob disease clinical phenotype with reactive depression, followed by cognitive impairment, akinetic-rigid Parkinsonism with pseudobulbar syndrome and gait impairment with motor apraxia, visuospatial disorientation, and evident frontal dysfunction features such as gras**, palmomental reflex and brisk perioral reflexes. She died at age 77.
Neuropathological findings showed: spongiform change in the patient’s cerebral cortex, striatum, thalamus and molecular layer of the cerebellum with proteinase K-sensitive synaptic-like, dot-like or target-like prion protein deposition in the cortex, thalamus and striatum; proteinase K-resistant prion protein in the same regions; and elongated plaque-like proteinase K-resistant prion protein in the molecular layer of the cerebellum. Molecular analysis of prion protein after proteinase K digestion revealed decreased signal intensity in immunoblot, a ladder-like protein pattern, and a 71% reduction of PrPSc signal relative to non-digested material. Her cerebellum showed a 2A prion protein type largely resistant to proteinase K. Genotype of polymorphism at codon 129 was valine homozygous.
Conclusion
Molecular ty** of prion protein along with clinical and neuropathological data revealed, to the best of our knowledge, the first case of the coexistence of different protease-sensitive prion proteins in the same patient in a rare case that did not fulfill the current clinical diagnostic criteria for either probable or possible sporadic Creutzfeldt–Jakob disease. This highlights the importance of molecular analyses of several brain regions in order to correctly diagnose rare and atypical prionopathies.
Similar content being viewed by others

Introduction
Prion diseases are fatal neurodegenerative disorders characterized by the accumulation of the abnormal isoform of the cellular prion protein (PrP). The main differences between the normal physiological and the abnormal pathologic protein are their physical and chemical properties, and specifically their detergent solubility, α-helix and β-sheet content in the secondary structure, and degree of resistance to protease digestion.
A novel form of sporadic prion disease, termed variably protease-sensitive prionopathy (PSPr or VPSPr), has recently been reported [5].
Neuropathology
Spongiform change was found in the upper (Figure 1A) and inner (Figure 1D) layers of the cerebral cortex, including frontal and temporal cortex, entorhinal cortex and subiculum. The striatum and thalamus showed moderate spongiform change. PrP immunoreactivity followed a synaptic pattern and was accompanied by dot-like (target-like according to the nomenclature of Zou et al. [6]) deposits, particularly in the deep cortical layers. PrP deposition was largely sensitive to PK treatment, except for the majority of target-like deposits that were PK-resistant (Figure 1B, 1C, 1E, 1F). Perineuronal PrP-immunoreactive deposition was absent.

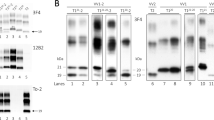
Immunohistological (A-L) and immunoblotting (M-N) results. A C: frontal cortex; D F: temporal cortex; G L: cerebellum. Spongiform change in the frontal (A) and temporal (D) cortex and molecular layer of the cerebellum (G, H) is accompanied by moderate neuronal loss in cortex (A, D) and torpedoes in the granular layer of the cerebellum (I). PrP-immunoreactive (PrP-ir) deposits are seen in the cerebral cortex and cerebellum (B, E, J). PrP-ir is largely reduced in the cerebral cortex after proteinase K (PK) treatment, except for small PrP-ir dots following a dot-like or target-like pattern (C, F). By contrast, PrP-ir in the molecular layer of the cerebellum, in the form of elongated plaque-like deposits, is preserved after PK treatment (K, L); PrP plaques in the granular layer are absent. Paraffin sections: A, D, G, H: hematoxylin and eosin staining; I: phosphorylated neurofilament immunohistochemistry; B, C, E, F, J L: PrP immunostaining (3F4 antibody) without (B, E, J) and with (C, F, K, L) PK treatment. A, D, G, J, K, L: × 200 (bar in L, 100μm); B, C, E, F, H, I: × 400 (bar in I, 50μm). PK was used according to the indications of the supplier: 1 drop of PK concentrate (DAKO, S2019) in 1.6mL of DAKO ChemMate TM PK diluent (S2032) for 15 minutes. M: Routine immunoblotting conditions (10% brain homogenate and final PK concentration of 440μg/mL) as described elsewhere [5] and five minutes of film exposure time. PK pretreated brain regions corresponded to occipital cortex (lane 1), putamen/globus pallidus (lane 2), cerebellum (lane 3), parietal cortex (lane 4), thalamus (lane 5), frontal cortex (lane 6), temporal cortex (lane 7), sporadic Creutzfeldt–Jakob disease (sCJD) VV2 reference case occipital cortex (lane 8). N: Immunoblotting of PK pretreated samples with less stringent conditions (TeSeE® Western Blot Kit, Bio-Rad) and detection with 3F4 antibody (Dako, dilution 1:3000) as previously described [5] at ten minutes film exposure time. Brain regions corresponded to occipital cortex (lane 1), cerebellum (lane 2), parietal cortex (lane 3), thalamus (lane 4), frontal cortex (lane 5), temporal cortex (lane 6), variably protease-sensitive prionopathy 129MV parietal cortex (lane 7) [5] and sCJD VV2 reference case frontal cortex (lane 8). Molecular weight standards are indicated in kDa: (M) SDS-PAGE Standards, broad range, Bio-Rad and (N) Precision Plus Protein Unstained Standards, Bio-Rad.
The cerebellum showed spongiform change in the molecular layer, moderate loss of Purkinje cells and occasional axonal torpedoes (Figure 1G–I). PrP immunohistochemistry showed elongated plaque-like deposits in the molecular layer which were resistant to PK pretreatment (Figure 1J–L). Plaque-like deposits in the granular layer of the cerebellum as are often seen in sCJD VV2 were absent.
Accompanying findings were neurofibrillary tangles in the entorhinal cortex, subiculum, hippocampus, inferior temporal cortex, and rarely in the frontal cortex, and β-amyloid plaques, diffuse and neuritic, in the cerebral cortex corresponding to Alzheimer’s disease-related changes in neuritic stages VB of Braak.
PRNP analysis
The entire PRNP open reading frame was amplified by polymerase chain reaction (PCR ) using genomic DNA extracted from fresh brain tissue. PCR was carried out in a final volume of 25μl with 45ng DNA, 0.1mM dNTP, 1mM MgCl2, 0.2μM of each primer (Forward: 5′-CCATTGCTATGCACTCATTC-3′; Reverse: 5′-CGGGACAAAGAGAGAAGAAA-3′), 0.2mg/ml bovine serum albumin BSA, 0.5 units of Taq Polymerase and 1x Taq buffer, under the following conditions: initial denaturation step at 95°C for ten minutes, 35 cycles of denaturation at 95°C for 45 seconds, annealing at 60°C for 30 seconds and elongation at 72°C for one minute, and a final elongation step at 72°C for ten minutes. Sequence analysis was done directly by automated sequencing. The patient was homozygous for valine at codon 129 of PRNP and no mutations, insertions or silent polymorphisms were found.
Biochemistry and western blotting
Routine immunoblot examinations [5] revealed lack of signal in all brain regions and a weak three band pattern of pathogenic PrP in the cerebellum corresponding to a 2A protein type (Figure 1M). The recent description of a case of protease-sensitive prionopathy 129MV and the use of technologies based on PrP detection without proteinase K treatment [5] allowed us to reanalyze this case. Immunoblotting under less restrictive conditions [5] and incubation with 3F4 antibody revealed a ladder-like PrP pattern in all brain regions excluding the cerebellum, which maintained the three band pattern typical of sCJD type 2A (Figure 1N). The evaluation of PrP PK sensitivity in the comparative ELISA showed a PrP mean signal detection rate decreased by 71% [5]. The highest values ranged from 72% to 78% for cortex, striatum and thalamus, and the lowest value of 43% corresponded to the cerebellum. This could partially explain the absence of protein signal in immunoblot in the aforementioned brain regions and the weak signal detected in the cerebellum.
Conclusion
The first 129VV protease-sensitive prionopathy series reported [1, 2, 4, 6]. However, a striking observation of this case was that PrP type in the cerebellum was reminiscent of sCJD type VV2 because PrP-resistant elongated deposits were present in the molecular layer plaques but absent in the granular layer. Reanalysis of different brain homogenates confirmed this finding. Co-occurrence of different PK-sensitivity PrP types in the same PSPr patient has, to the best of our knowledge, not yet been reported [1, 2, 4, 6], even though coexistence of different classical PK-resistant PrP forms is a common finding in other prion diseases [7–15]. This observation constitutes a novel finding which broadens the spectrum of molecular and clinicopathological features of spongiform encephalopathies.
Consent
Written informed consent was obtained from the patient's next-of-kin for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Gambetti P, Dong Z, Yuan J, **ao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D: A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008, 63: 697-708. 10.1002/ana.21420.
Head MW, Knight R, Zeidler M, Yull H, Barlow A, Ironside JW: A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol. 2009, 35: 628-632.
Head MW, Lowrie S, Chohan G, Knight R, Scoones DJ, Ironside JW: Variably protease-sensitive prionopathy in a PRNP codon 129 heterozygous UK patient with co-existing tau, alpha synuclein and Abeta pathology. Acta Neuropathol. 2010, 120: 821-823. 10.1007/s00401-010-0766-y.
Jansen C, Head MW, van Gool WA, Baas F, Yull H, Ironside JW, Rozemuller AJ: The first case of protease-sensitive prionopathy (PSPr) in The Netherlands: a patient with an unusual GSS-like clinical phenotype. J Neurol Neurosurg Psychiatry. 2010, 81: 1052-1055. 10.1136/jnnp.2009.175646.
Rodriguez-Martinez AB, Garrido JM, Zarranz JJ, Arteagoitia JM, de Pancorbo MM, Atares B, Bilbao MJ, Ferrer I, Juste RA: A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: case report. BMC Neurol. 2010, 10: 99-10.1186/1471-2377-10-99.
Zou WQ, Puoti G, **ao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S: Variably protease-sensitive prionopathy: A new sporadic disease of the prion protein. Ann Neurol. 2010, 68: 162-172. 10.1002/ana.22094.
Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, Mackenzie J, Knight R: Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991–2002. Ann Neurol. 2004, 55: 851-859. 10.1002/ana.20127.
Kovacs GG, Head MW, Hegyi I, Bunn T, Flicker H, Hainfellner JA, McCardle L, Laszlo L, Jarius C, Ironside JW: Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol. 2002, 12: 1-11.
Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ: Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology. 2005, 65: 113-118. 10.1212/01.wnl.0000167188.65787.a0.
Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P, Kretzschmar HA, Parchi P: A refined method for molecular ty** reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest. 2007, 87: 1103-1112. 10.1038/labinvest.3700676.
Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, Zerr I, Roncaroli F, Cras P, Ghetti B: Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009, 118: 659-671. 10.1007/s00401-009-0585-1.
Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A: Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005, 4: 805-814. 10.1016/S1474-4422(05)70225-8.
Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F: Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology. 1999, 53: 2173-2176. 10.1212/WNL.53.9.2173.
Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M: Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2006, 3: e14-10.1371/journal.pmed.0030014.
Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, Ironside JW, Haik S, Basset-Leobon C, Lacroux C: Beyond PrP res type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008, 4: e1000029. 10.1371/journal.ppat.1000029.
Acknowledgments
This work was supported by a Support to Health Research grant of the Planning and Arranging Director of the Department of Health of the Basque Government (Grant #2006111037). We thank the family of the patient for granting permission for publication of this case.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AR carried out the molecular and genetic analysis. JA organized and managed the necropsy and the epidemiological information. AL, IF, NV and BA collected and evaluated clinical and pathological information and samples. AR, AL, IF, RJ and JZ participated in the drafting of the manuscript and critically discussed and wrote specific parts of it. AR, RJ and JG conceived the study. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rodríguez-Martínez, A.B., de Munain, A.L., Ferrer, I. et al. Coexistence of protease sensitive and resistant prion protein in 129VV homozygous sporadic Creutzfeldt–Jakob disease: a case report. J Med Case Reports 6, 348 (2012). https://doi.org/10.1186/1752-1947-6-348
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-1947-6-348