
Abstract
Background
The eastern oyster, Crassostrea virginica, is a euryhaline species that can thrive across a wide range of salinities (5-35). As with all estuarine species, individual oysters must be able to regulate their osmotic balance in response to constant temporal variation in salinity. At the population level, recurrent viability selection may be an additional mechanism sha** adaptive osmoregulatory phenotypes at the margins of oyster salinity tolerance. To identify candidate genes for osmoregulation, we sequenced, assembled, and annotated the transcriptome of wild juvenile eastern oysters from ‘high’ and ‘low’ salinity regimes. Annotations and candidates were mostly based on the Pacific oyster (Crassostrea gigas) genome sequence so osmoregulatory relevance in C. virginica was explored by testing functional enrichment of genes showing spatially discrete patterns of expression and by quantifying coding sequence divergence.
Results
The assembly of sequence reads and permissive clustering of potentially oversplit alleles resulted in 98,729 reftigs (contigs and singletons). Of these, 50,736 were annotated with 9,307 belonging to a set of candidate osmoregulatory genes identified from the C. gigas genome. A total of 218,777 SNPs (0.0185 SNPs/bp) were identified in annotated reftigs of C. virginica. Amino acid divergence between translations of C. virginica annotated reftigs and C. gigas coding sequence averaged 23.2 % with an average dN/dS ratio of 0.074, suggesting purifying selection on protein sequences. The high and low salinity source oysters each expressed a subset of genes unique to that group, and the functions for these annotated genes were consistent with known molecular mechanisms for osmotic regulation in molluscs.
Conclusions
Most of the osmoregulatory gene candidates experimentally identified in C. gigas are present in this C. virginica transcriptome. In general these congeners show coding sequence divergence too high to make the C. gigas genome a useful reference for C. virginica bioinformatics. However, strong purifying selection is characteristic of the osmoregulatory candidates so functional annotations are likely to correspond. An initial examination of C. virginica presence/absence expression patterns across the salinity gradient in a single estuary suggests that many of these candidates have expression patterns that co-vary with salinity, consistent with osmoregulatory function in C. virginica.
Similar content being viewed by others

Background
The eastern oyster (Crassostrea virginica) builds reefs that support productive estuarine communities and provide important ecosystem services [1, 2]. However, overfishing, disease pressure, and environmental stress have led to the loss of approximately 90% of biomass across the eastern oyster’s home range since the early 1900’s [3–5]. Two important topics in oyster biology and restoration are the mechanisms by which oysters respond to stress [6, 7] and the ability of oyster populations to either acclimate to stress through phenotypic plasticity or adapt via selection. The majority of work on eastern oysters has focused on immune response to pathogens [8–11] with a few observational studies on other environmental stressors [6, 12]. Spatial and temporal variation in salinity is a given for estuaries, and phenotypic buffering of cell volume through osmolyte control is an essential adaptation for all organisms that live there.
Eastern oysters are found along salinity gradients ranging from near freshwater conditions (salinity of 5) to oceanic salinities (salinity of 35) [13–15]. Their greatest abundance is typically at intermediate salinities, with the adult physiological optimum posited to be as narrow as salinities of 15–18 [15]. At the margins of this environmental envelope, recent results suggest that post-settlement viability selection is one important process for sustaining adult populations [16]. While the genes involved in osmoregulation have not been well characterized in the eastern oyster, recent studies on the Pacific oyster (Crassostrea gigas) [29]. Particularly in highly polymorphic species such as oysters, alleles will often assemble into separate contigs or remain as singletons. This redundancy complicates downstream applications of the resulting transcriptome, such as gene expression analyses [30], because reads from some single copy genes will not map uniquely within the transcriptome and may be discarded. We assumed that the reftigs from the Newbler assembly included many oversplit loci, so to improve the transcriptome we consolidated redundant reftigs by clustering. Cd-hit [31] was used to cluster reftigs with various sequence identity thresholds ranging from permissive clustering at 80% to conservative clustering at 99% using a k-mer word size of 5 to 10 increasing incrementally with the threshold (e.g. word size 5 with threshold 80%).
Several approaches were used to evaluate whether clustering improved the transcriptome. For the 80% and 95% clustering results we compared statistics bearing on transcriptome quality including the percent of reftigs that were annotated, the distribution of annotation between contigs and singletons, and the number of osmoregulatory candidates (identified in C. gigas) recovered.
Additionally, we evaluated the two clustering results by comparing the proportion of Illumina reads from a barcoded individual that uniquely mapped to annotated reftigs based on a pilot RNA-seq experiment. The barcoded individual was from an oyster reef in Delaware Bay with a salinity regime ranging from 6.5 to 14.5. The mRNA from 30 mg of gill tissue was extracted using Dynabeads® mRNA DIRECT™ kit (Life Technologies). The library was prepared with NEBNext® mRNA Library Pep Reagent Set for Illumina® (New England BioLabs Inc.). The library constituted 16.25% of a single 100 bp Hi-Seq Illumina lane and was sequenced at the Biotechnology Resource Center Genomics Facility of Cornell University. The resulting reads were trimmed following the same procedure as the 454 reads, and any remaining adapters were clipped using fastx_clipper (FASTXToolkit). The remaining reads were then mapped to the annotated reftigs using BWA [32] with a mismatch edit distance of 0.005 and SAMtools [33] with only uniquely mapped reads retained.
Annotation
To annotate the de novo C. virginica transcriptome assembly, reftigs were compared to NCBI's non-redundant (nr) protein sequence database that included the annotated proteins deduced from the C. gigas genome (May 2013), plus the Swiss-Prot and TrEMBL databases from the Uniprot protein knowledge base, using the BLASTx algorithm with an e-value cut-off of 10-5. Gene Ontology (GO; http://www.geneontology.org) annotation was retrieved from Uniprot. The annotated and unannotated reftigs were then compared with respect to the proportion of contigs and singletons as well as GC content in order to explore if unannotated reftigs may represent non-oyster contamination in the 454 sequences. The number of unique genes represented by the transcriptome was then identified by grou** reftigs that shared the same GenBank gene identifier.
We considered genes as osmoregulatory candidates if they were included in the 1,241 annotated genes found to be differentially expressed in C. gigas adults in response to six different salinity treatments when compared to a control salinity of 30 Table S21 in [34]. Genes that were unique to one of the two populations were identified as “asymmetric.” Results from enrichment tests were depicted in the context of the hierarchical structure of gene ontology terms in order to visualize the degree of functional integration among the most significantly enriched genes.
Sequence comparisons with C. gigas
Simple sequence repeats and low complexity regions of the annotated reftigs were masked with RepeatMasker [35], using the rmblast search engine. Reftigs with masked regions were removed from analysis. The coding sequence reading frame for each remaining reftig was then predicted using ESTscan [36]. ESTscan was trained using the EMBL, RefSeq and UniGene clusters from the mollusk Aplysia californica, the most closely related species for which a full set of references were available at the time of this study. The matrices from this training were then used to predict coding sequences for the reftigs using a hidden Markov model [36]. The predicted coding sequence for each reftig was then used to analyze sequence divergence from C. gigas.
A local directory of C. gigas coding sequence for predicted proteins from the C. gigas genome was downloaded from http://gigadb.org/dataset/view/id/100030/sort/size and clustered with Cd-hit using the same parameters as for reftig clustering (sequence identity threshold = 0.8, word size = 5). Coding sequences that clustered were assumed to be paralogs and removed from analysis to reduce the bias that would occur with comparison of paralogs between C. gigas and C. virginica. The C. virginica reftigs were then compared against the C. gigas coding sequences using tBLASTx with intron linking disabled and an e-value cutoff of 10-5. Best hits were interpreted as putative ortholog pairs for analysis. Ortholog pairs were then run through a custom pipeline to align sequences using ClustalW [37] and calculate dN/dS ratios using the codeml function of paml [38]. The distribution of dN/dS values relative to ClustalW alignment length was evaluated before choosing to remove alignments less than 60% of the total reftig length.
SNP discovery
The mapped 454 reads from both population samples were combined and aligned against the masked, annotated reftigs with mpileup of SAMtools [33]. SNPs were then identified using SNAPE-pooled [39] with a base quality average of 37 or greater, theta of 0.01, divergence of 0.1, flat prior and folded spectrum, and the SNP density for each contig was calculated.
Results and discussion
Assembly and clustering results
A total of 1,256,652 raw 454 reads included 718,009 from the high salinity population and 538,553 from the low salinity population. The raw reads are available through the National Center for Biotechnology Information Short Read Archive under accession numbers SRS502377 for the high salinity population and SRS502378 for the low salinity population. After trimming and filtering, 1,182,107 reads remained and were assembled into 28,939 contigs that contained 86.7% of the reads. The 128,083 unassembled reads were designated as singletons and included in further analysis. The combined contig and singleton set consisted of 157,022 reftigs. The assembly size for contigs alone was approximately 18,202,631 nucleotide bases, similar to other molluscan transcriptome assemblies based on 454 sequences (Table 1), and had an average contig length of 629.1 bases (N50 = 500 bases) and maximum contig length of 7,512 bases. The total transcriptome (contigs and singletons, 157,022 reftigs) was 51,918,466 nucleotides with an average length of 453.0 bases (N50 = 381 bases).
Given the high degree of polymorphism in oysters [17], clustering of assembled reftigs was explored as a method of consolidating alleles that remained apart after assembly. Consolidating alleles is an important consideration before using a transcriptome assembly as a reference for RNAseq expression analyses because oversplit alleles in the assembly will decrease the number of reads that map uniquely. A total of 136,000 reftigs (86.7%) were longer than 200 bp and used for clustering at different sequence identity thresholds. Comparing the change in total reftig number resulting from increasingly permissive clustering, the rate of reftig consolidation was initially rapid based on thresholds from 99% to 95%; then the rate of change slowed and was nearly constant between 95% and 80% (Figure 2). As the sequence identity threshold decreased, the ratio of contigs to singletons increased as expected if singletons were being clustered with greater frequency than contigs (Table 2).

Collapsing of reftigs with decreasing sequence identity threshold in Cd-hit. As sequence identity threshold decreases in Cd-hit clustering, the number of reftigs decreases. The rate of reftig consolidation is highest from 1 to 0.95 but remains consistent from 0.95 to 0.90.
Transcriptomes resulting from both the 95% and 80% sequence identity thresholds were annotated for comparison. Both transcriptomes had a similar percentage of reftigs successfully annotated, with a similar distribution of contigs and singletons. Likewise, 1,014 osmoregulatory candidates (see below) were obtained with the 95% threshold and this dropped by only seven candidate genes at the 80% threshold (Table 2). The large percentage (99.4%) of candidate genes that remain in the transcriptome at the 80% threshold compared to the 95% threshold suggests that any potential paralog clustering resulted in a minimal loss in the number of uniquely annotated genes, particularly osmoregulatory candidates.
We mapped 100 bp Illumina RNAseq reads from a single individual to the 95% and 80% transcriptomes to test whether oversplit alleles were consolidated by clustering. Relative to the 95% transcriptome, the 80% transcriptome had a higher percentage of annotated reftigs with mapped reads, but the effect was small (Table 2). In terms of the proportion of Illumina reads that mapped, two percent more reads mapped uniquely to the 80% transcriptome than the 95% transcriptome. The increase in the percent of uniquely mapped reads in the 80% transcriptome suggests that consolidation of allelic reftigs was achieved by clustering, resulting in more reads map** uniquely. Based on these results the 80% threshold transcriptome was chosen for further analysis.
In one cluster examined in more detail, an original contig annotated as Heat Shock 70 kDA Protein 12 was ultimately clustered with three singletons. Two singletons clustered at the 95% threshold. The third singleton (330 bp) was unannotated at the 95% threshold. With a similarity of 84.55% estimated by Cd-hit, it was clustered with the contig and the other two singletons at the 80% threshold. An alignment between this third singleton and the original contig showed five indels ranging in size from 1 to 17 bp and two polymorphisms as the cause for the 84.55% sequence identity. We suspect these indels represent 454 sequencing error because they were partially shared across the three singletons, most of them would disrupt the reading frame, and they occurred within simple nucleotide repeats and low complexity sequence. Some of the SNPs present in the singletons may also be sequencing error but not obviously so; most were not adjacent to indels and they were already represented in the original contig. Therefore, clustering provided two distinct transcriptome improvements: (1) oversplit alleles were consolidated, facilitating downstream map** to the transcriptome for RNAseq expression analysis, and (2) more of the 454 sequencing coverage was used to call SNPs.
The optimum balance between consolidating oversplit alleles and clustering paralogs or sequence errors is impossible to know because it depends on the distribution of allelic sequence differences relative to paralog differences in any particular species as well as the sequencing error rate. The comparative approach used here was ad hoc and took advantage of computational efficiencies when clustering consensus sequences from an assembly rather than exploring parameter values in separate assemblies. When no reference genome is available, this comparative empirical approach can be a valuable method of improving transcriptome quality.
Annotation results
The BLASTx search against multiple databases provided annotation for 50,736 reftigs (51.4%) representing 20,249 unique GenBank accessions and 16,392 distinct putative proteins that we will refer to as genes. Only 0.05% of the annotations were achieved with a database other than GenBank nr (Figure 3). Reftigs that did not have a BLASTx match with an e-value smaller than 10-5 from any database was designated as unannotated.

Number of reftigs annotated by each database out of 50,736 total reftigs. The Genbank nr database supplied 99.95% of annotations.
Of 16,392 distinct genes, 8,161 are represented by a single reftig. The number of reftigs per gene ranges from 1 to 470 with only 13 genes represented by 100 or more reftigs. Gene duplication and large gene families, particularly in the C. gigas genome from which 89% of our annotations were identified, are the primary reasons for the large number of reftigs per “gene”. For example, the 456 reftigs identified as the gene “tripartite motif-containing protein 2” from C. gigas were annotated from 201 unique GenBank accessions. For C. gigas, these different GenBank accessions represent different coding sequence locations within the genome. Therefore, we define a “gene” here as a protein product, which often represents large gene families.
Gene Ontology (GO) terms were assigned to 36,924 of the annotated reftigs, representing 11,583 putative genes, based on sequence similarity to known proteins in the UniProt databases. Annotated reftig sequences have been archived and are accessible through FigShare (http://dx.doi.org/10.6084/m9.figshare.873865) [51].
A consequence of whole animal extractions and normalizing the libraries was the increased potential to sequence non-oyster transcripts, such as bacteria and algae. Singletons made up ~74% of the reftigs in the full (annotated and unannotated) 80% transcriptome and less than half of the singletons were successfully annotated. Other studies using 454 sequencing also have described singletons as comprising a large proportion of their transcriptome (e.g. 81.7% [54].
Fifty-nine osmoregulatory candidate genes were identified from only the high salinity population and 56 were identified from only low salinity, together representing 11.4% of the candidate genes. These asymmetrically expressed candidate genes were mostly cases with 1x coverage (1:0 asymmetry), but 3 genes (2.6%) had an asymmetry ratio of 5:0 or greater.
Ignoring candidate status, 4,053 of the 16,392 annotated C. virginica genes (24.7%) were identified from only one of the two populations. Of these, 2,185 were found only in the high-salinity population, including 1,431 genes (8.7%) with 1:0 asymmetry and 74 genes (0.5%) with 5:0 or greater asymmetry. An additional 1,868 genes were found only in the low-salinity population, including 1,355 (8.3%) with a 1:0 asymmetry and 31 genes (0.2%) with 5:0 or greater asymmetry. A total of 105 genes (2.6%) from the two populations had 5:0 or greater asymmetry.
The fact that 24.7% of all genes showed asymmetry, while only 11% of osmoregulatory candidates did so, suggests that there may be many biological processes leading to population-specific expression in addition to the stochasticity expected with low-expression genes. Also, given that buffering against osmotic stress is a chronic physiological need for oysters, lower asymmetry among osmoregulatory candidates might reflect the proportion of genes within this functional category that have constitutive expression across salinities.
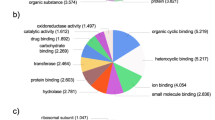
It is difficult to know how much asymmetry to expect by chance for genes with a given level of expression in normalized libraries. However, enrichment of functional categories within the set of population-specific genes is not expected from stochastic variation in library normalization or read coverage. Our prediction was that among asymmetric genes, annotations related to osmoregulatory function should be the most highly enriched relative to the frequency of functional ontologies in the overall annotated transcriptome. We initially built a frame of reference by testing for functional enrichment among the entire set of osmoregulatory candidate genes in C. virginica and found 12 cellular component GO terms and 86 molecular function GO terms significantly enriched compared to the complete annotated gene set (Figures 5 and 6, Additional file 2: Tables S2 and S3). For cellular components, the ‘extracellular region’ , ‘plasma membrane’ and ‘membrane’ components were among the significantly enriched terms (Figure 5 and Additional file 2: Table S2). At the level of molecular function, ‘catalytic’ activities, ‘binding’ functions, ‘electron carrier’ activities, ‘transporter’ activities and ‘molecular transducer’ activities were among the significantly enriched terms (Figure 6, Additional file 2: Table S3). These enriched GO terms serve to functionally characterize the osmoregulatory candidates on the whole and therefore might be indicators of osmoregulatory function in additional enrichment tests when found in concert.

Distribution of level 3 Cellular Component GO terms for the osmoregulatory candidate genes. Black bars indicate the terms or the parents of GO terms that are significantly enriched in the osmoregulatory candidate genes compared to the complete set of annotated genes.

Distribution of level 3 Molecular Function GO terms for the osmoregulatory candidate genes. Black bars indicate the terms or the parents of GO terms that are significantly enriched in the osmoregulatory candidate genes compared to the complete set of annotated genes.
As predicted, the overall group of asymmetric genes (24.7% of all genes) showed significantly enriched ontologies relating to osmoregulatory function, as indicated by similarities with enriched GO terms in the total set of osmoregulatory candidate genes. Interestingly, the enriched GO terms were only partially overlap** in the low versus high salinity population, and relative enrichment magnitudes shifted among GO terms. In the low salinity population at the level of cellular components, the strongest result among 12 significantly enriched ontologies included ‘integral to membrane’ (GO:0016021, p = 0.00037) and ‘intrinsic to membrane’ (GO:0031224, p = 0.00058), two ontologies nested within ‘membrane’ components, the level-three GO term enriched among osmoregulatory candidates (Additional file 2: Table S4). Transmembrane channels are important in maintaining cell volume in response to hypoosmotic stress. For example, Ca2+ channels are upregulated in hypoosmotic stress and osmolytes such as taurine are taken up through high affinity transport systems that may involve transmembrane proteins [18]. Additional terms such as ‘cell periphery’ (GO:0071944, p = 0.0033), ‘plasma membrane part’ (GO:0044459, p = 0.00624) and ‘plasma membrane’ (GO:0005886, p = 0.00815) were terms significantly enriched both in the asymmetric low salinity genes and the full candidate gene set (Additional file 2: Tables S2 and S4). In general, however, the cellular component terms most strongly enriched in the full set of osmoregulatory candidates, extracellular region and its ‘children’ terms, were not enriched in genes expressed solely at low salinity in C. virginica.
In contrast to the low population, the most significant functional enrichment at the level of cellular components in the high population was ‘extracellular region’ (GO:0005576, p = 6.4e-08). This term refers to the gene products that are secreted from the cell but retained in the interstitial fluid or hemolymph, and it was also the most significantly enriched for the full osmoregulatory candidate gene set (Additional file 2: Table S2, Figure 5). While this parent GO term had the highest level of enrichment among the C. gigas genes experimentally associated with salinity treatments [19] (Additional file 2: Table S2), it is also likely to include immune response genes responding to the larger disease burden found in oysters from high salinity [19, 55]. Several additional GO terms were significantly enriched both in the asymmetric high salinity genes and in the full candidate gene set including ‘intrinsic to membrane’ (GO:0031224, p = 6.4e-06) and ‘plasma membrane’ (GO:0005886, p = 1.3e-05) (Additional file 2: Tables S2 and S5).
At the molecular functions GO level, both the high and low salinity populations showed the strongest significant enrichments related to DNA replication and transcription/translation (Additional file 2: Tables S6 and S7). For the low salinity population, many of the other significantly enriched molecular function ontologies (Additional file 2: Table S6) were ‘children’ terms of those significantly enriched both for osmoregulatory genes and for unique low salinity genes (Additional file 2: Tables S3 and S6, Figure 6). For example, ‘G-protein coupled receptor activity’ (GO:0004930, p = 6.6e-07) is a ‘child’ term of ‘receptor activity’ and ‘aspartic-type peptidase activity’ (GO:0004190, p = 2.2e-06) is a ‘child’ term of ‘hydrolase activity.’ These enriched functions match predictions that the phosphorylation of plasma membrane proteins and the hydrolysis of peptides are part of the physiological response to osmotic stress. For the high salinity population, significant enrichment was found for potential osmoregulatory terms related to ‘substrate-specific transporter’ and ‘transmembrane transporter’ activities such as ‘receptor activity’ (GO:0004872, p = 7.8e-06), ‘gated channel activity’ (GO:0022839, p = 0.00022), and ‘ion gated channel activity’ (GO:0022839, p = 0.0022) (Additional file 2: Table S7). These enrichment results are consistent with expectations for differential expression of osmoregulatory genes by juvenile eastern oysters from different salinity regimes. Furthermore, it confirms the functional relevance in C. virginica of osmoregulatory candidates identified in C. gigas.
At the level of reftigs, rather than genes, among those with annotations linked to osmoregulatory function in C. gigas (9703 reftigs), 57.3% showed expression in only one of the two populations. This high frequency of asymmetry is in striking contrast to the 11% asymmetry measured at the gene level among osmoregulatory candidates. One possible explanation for this pattern is that asymmetric reftigs represent differentially expressed splice variants of genes expressed by both populations. This hypothesis will be testable with the benefit of this transcriptome as a reference for RNA-seq analyses.
SNP Discovery and dN/dS ratio with C. gigas
The transcriptome we present here provides the most comprehensive estimate of polymorphism to date for C. virginica. Among 13,108 annotated contigs, there was 12,355,575 bp of aligned sequence within which 218,777 SNPs were identified. Average SNP density was 0.0185 per base pair with a standard deviation of 0.0238 (Figure 7). Ninety percent of contigs had at least one predicted SNP. This SNP density falls within the range previously reported for the eastern oyster. Quilang et al. [56] found a rate of 0.0059 SNPs/bp from 4,688 EST sequences. In contrast, Zhang and Guo [57] estimated 0.042 SNPs/bp based on resequencing 6.8 kb of ESTs. Similarly, a single gene study of serine protease inhibitor reported an overall SNP frequency of 0.044/bp [58]. For comparison, SNP density averaged across all exons in wild-caught C. gigas was 0.0102 per bp [17]. Our finding is therefore consistent with previous estimates of nucleotide heterozygosity in C. virginica and tentatively supports the contention that this species is more polymorphic than C. gigas [57].

The distribution of SNP density per base pair within annotated contigs from the 80% clustered transcriptome.
Quantifying genomic patterns of divergence between C. virginica and C. gigas can help assess the relevance of discoveries in one species with respect to the other. Also, the ratio of substitution rates at nonsynonymous and synonymous sites can help to identify genes undergoing positive selection. After various filtering steps to remove potential artifacts and paralog gene pairs, 26,102 annotated reftigs from C. virginica were paired with an ortholog from C. gigas. Estimates for the number of nonsynonymous substitutions per nonsynonymous site ranged from near 0 to 0.012/bp per ortholog gene pair (Figure 8A) while the number of synonymous substitutions per synonymous site ranged from 0.0003 to 0.66/bp (Figure 8B). The mean dN/dS ratio of 0.074 (SD = 0.066, Figure 8C) indicates a pervasive role for purifying selection maintaining similar amino acid sequences. The mean protein similarity was 76.8%, and the mean nucleotide similarity was 74.2%. It is possible that these divergence estimates between the oyster congeners are biased downward because filtering steps (see Methods) inevitably removed some more divergent ortholog pairs.

Distribution of (A) dN, (B) dS and (C) dN/dS ratio values. dN and dS show similar distribution shapes with the number of synonymous substitutions much larger than the number of nonsynonymous substitutions. Most ortholog pairs had a dN/dS ratio below 0.2 indicating a strong role for purifying selection on oyster peptide sequences.
This degree of purifying selection provides some confidence that functional candidate genes identified in C. gigas will often be applicable to C. virginica, at least as a starting point. At the same time, transcriptomes in these two species are probably too diverged to expect C. gigas genomic reference sequences to help with C. virginica bioinformatics. A simulation study by Vijay et al. [59] demonstrated that reference genomes with average nucleotide sequence divergence up to 15% can help improve transcriptome assemblies while with greater divergence there was no improvement over a de novo assembly. Similarly, the potential for a heterologous reference genome to provide improved RNA-seq analyses, relative to a de novo transcriptome assembly, was determined to be at nucleotide sequence divergences less than 15% [59].
Conclusions
The goal of our study was to assemble and annotate the C. virginica transcriptome with particular focus on potential osmoregulatory genes. Largely with the benefit of the Pacific oyster genome, we assigned provisional annotations to 50,736 reftigs representing over 16,000 putative proteins. More than 80% of the osmoregulatory gene candidates identified in C. gigas experiments with adults were identified here in wild juvenile samples from different salinities. The low dN/dS between C. virginica and C. gigas indicates purifying selection in the coding regions of orthologous genes and provides justification that genes identified as osmoregulatory in C. gigas are likely to maintain the same function in C. virginica. Even stronger justification is reported for a subset of osmoregulatory candidates that were expressed in only one of the two different salinity populations. Genes with an asymmetric expression pattern across the salinity gradient were significantly enriched for functions that may be related to osmoregulation, consistent with these genes having osmoregulatory functions in C. virginica.
Additionally, we have demonstrated that permissive clustering of contig and singleton sequences may improve downstream applications of assembled transcriptomes. In some de novo transcriptome assembly studies, the singleton reftigs are discarded and only the contigs are analyzed. Such a stringent filter, if applied here to C. virginica, would have eliminated 37,717 singletons that were successfully annotated. The goal of clustering is to keep the singletons and reduce redundancy across reftigs that can result from de novo assembly challenges due to factors such as sequencing error and high levels of polymorphism. Several studies employing programs such as Cd-hit to cluster sequences based on similarity used a threshold of 95% similarity [7, 49]. We explored a range of threshold values from 99% down to the lower limit of the algorithm at 80%. The improvement in uniquely mapped reads may be beneficial for downstream applications, depending on experimental goals. For RNA-seq experiments, a greater number of uniquely mapped reads means that a greater percentage of data can be retained for the estimation of expression. Future development of these clustering procedures should focus on evaluating trade-offs, particularly with respect to the incorporation of sequencing error at more permissive clustering thresholds.
Finally, we have provided a valuable set of resources for eastern oyster research. We have annotated 50,736 reftigs, doubling the 48,183 C. virginica transcriptome contig sequences provided by Zhang et al. [7]. After careful filtering of these reftigs, we identified 218,777 candidate SNPs for use in genetic map** or for population analyses. The 1,007 candidate genes for osmoregulation identified here will provide a reference for future studies on the molecular basis of osmoregulation in C. virginica, phenotypically plastic responses to salinity stress, and patterns of selective differentiation across heterogeneous environments.
Availability of supporting data
The data sets supporting the results of this article are available in the National Center for Biotechnology Information Short Read Archive, accession numbers SRS502377 (http://www.ncbi.nlm.nih.gov/biosample/SRS502377) and SRS502378 (http://www.ncbi.nlm.nih.gov/biosample/SRS502378), and in FigShare (http://dx.doi.org/10.6084/m9.figshare.873865).
References
Coen LD, Luckenbach MW, Breitburg DL: The role of oyster reefs as essential fish habitat: a review of current knowledge and some new perspectives. Am Fish Soc Symp. 1999, 34: 303-307.
Coen LD, Brumbaugh RD, Bushek D, Grizzle R, Luckenback MW, Posey MH, Powers SP, Tolley SG: Ecosystem services related to oyster restoration. Mar Ecol Prog Ser. 2007, 341: 303-307.
Zu Ermgassen PSE, Spalding MD, Grizzle RE, Brumbaugh RD: Quantifying the loss of a marine ecosystem service: Filtration by the eastern oyster in US estuaries. Estuar Coasts. 2013, 36: 36-43.
Beck MW, Brumbaugh RD, Airoldi L, Carranza A, Coen LD, Crawford C, Defeo O, Edgar GJ, Hancock B, Kay M, Lenihan HS, Luckenbach MW, Toropova CL, Zhang G, Guo X: Oyster reefs at risk and recommendations for conservation, restoration and management. Bioscience. 2011, 61: 107-116.
Willberg MJ, Livings ME, Barkman JS, Morris BT, Robinson JM: Overfishing, disease, habitat loss, and potential extirpation of oysters in the upper Chesapeake Bay. Mar Ecol Prog Ser. 2011, 436: 131-144.
Chapman RW, Mancia A, Beal M, Veloso A, Rathburn C, Blair A, Holland AF, Warr GW, Didinato G, Sokolova IM, Wirth EF, Duffy E, Sanger D: The transcriptomic response of eastern oyster, Crassostrea virginica, to environmental conditions. Mol Ecol. 2011, 20 (7): 1431-1449.
Zhang L, Li L, Zhu Y, Zhang G, Guo X: Transcriptome analysis reveals a rich gene set related to innate immunity in the eastern oyster (Crassostrea virginica). Mar Biotechnol. 2013, 2013: 1-17.
Goedken M, Morsey B, Sunila I, de Guise S: Immunomodulation of Crassostrea gigas and Crassostrea virginica cellular defense mechanisms by Perkinsus marinus. J Shellfish Res. 2005, 24 (2): 487-496.
Tanguy A, Guo X, Ford SE: Discovery of genes expressed in response to Perkinsus marinus challenge in Eastern (Crassostrea virginica) and Pacific (Crassostrea gigas) oysters. Gene. 2004, 338 (1): 121-131.
Jenny MJ, Ringwood AH, Lacy ER, Lewitus AJ, Kempton JW, Gross PS, Warr GW, Chapman RW: Potential indicators of stress response identified by expressed sequence tag analysis of hemocytes and embryos from the American oyster, Crassostrea virginica. Mar Biotechnol. 2002, 4 (1): 81-93.
Anderson RS, Burreson EM, Paynter KT: Defense responses of hemocytes withdrawn from Crassostrea virginica infected with Perkinsus marinus. J Invertebr Pathol. 1995, 66: 82-89.
Ivanina AV, Taylor C, Sokolova IM: Effects of elevated temperature and cadmium exposure on stress protein response in eastern oysters Crassostrea virginica (Gmelin). Aquat Toxicol. 2009, 91 (3): 245-254.
Loosanoff VL: Behavior of oysters in waters of low salinity. Proceed Natl Shellfisheries Assoc. 1952, 1953: 135-151.
Shaw WN: Oyster setting in two adjacent tributaries of Chesapeake Bay. ASB Bull. 1966, 13 (2): 45-
Shumway SE: Natural environmental factors. The Eastern Oyster Crassostrea virginica. Edited by: Kennedy VS, Newell RIE, Eble AF. 1996, College Park, Maryland: Maryland Sea Grant College Publication, 467-513.
Eierman LE, Hare MP: Survival of oyster larvae in different salinities depends on source population within an estuary. J Exp Mar Biol Ecol. 2013, 449: 61-68.
Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H, **ong Z, Que H, **e Y, Holland PWH, Paps J, Zhu Y, Wu F, Chen Y, Wang J, Peng C, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z, Zhu Q, Feng Y, Mount A, Hedgecock D, et al: The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012, 490: 49-54.
Meng J, Zhu Q, Zhang L, Li C, Li L, She Z, Huang B, Zhang G: Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS One. 2013, 8 (3): e58563-doi:10.1371/journal.pone.0058563
Zhao Z, Yu H, Kong K, Li Q: Transcriptomic responses to salinity stress in the Pacific oyster Crassostrea gigas. PLoS One. 2012, 7 (9): e46244-doi:10.1371/journal.pone.0046244
Evans DH: Osmotic and Ionic Regulation: Cells and Animals. 2009, Boca Raton: CRC Press
Pierce SK: Invertebrate cell volume control mechanisms: A coordinated use of intracellular amino acids and inorganic ions as osmotic solute. Biol Bull. 1982, 163: 405-419. http://dx.doi.org/10.2307/1541452,
Hosoi M, Kubota S, Toyohora M, Toyohora H, Hayashi I: Effect of salinity change on free amino acid content in Pacific oyster. Fish Sci. 2003, 69 (2): 395-400.
Pierce SK, Rowland-Faux LM, O’Brien SM: Different salinity tolerance mechanisms in Atlantic and Chesapeake Bay conspecific osyters: glycine betaine and amino acid pool variations. Mar Biol. 1992, 113: 107-115.
Yancey PH: Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol. 2005, 208: 2819-2830.
Toyohora H, Yoshida M, Hosoi M, Hayashi I: Expression of taurine transporter in response to hupo-osmotic stress in the mantle of Mediterranean blue mussel. Fish Sci. 2005, 71: 356-360.
Hosoi M, Shinzato C, Masaya T, Hosoi-Tanabe S, Sawada H, Terasawa E, Toyohara H: Taurine transporter function from the giant Pacific oyster Crassostrea gigas: function and expression in response to hyper- and hypo-osmotic stress. Fish Sci. 2007, 73: 385-394.
Ren J, Liu X, Jiang F, Guo X, Liu B: Unusual conservation of mitochondrial gene order in Crassostrea oysters: evidence for recent speciation in Asia. BMC Evol Biol. 2010, 10: 394-
Lambert JD, Chan XY, Spiecker B, Sweet HC: Characterizing the embryonic transcriptome of the snail Ilyanassa. Integr Comp Biol. 2010, 50 (5): 768-777.
Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV: Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009, 10: 219-
Wolf JBW, Bayer T, Haubold B, Schilhabel M, Rosenstiel P, Tautz D: Nucleotide divergence vs. gene expression differentiation: comparative transcriptome sequencing in natural isolates from the carrion crow and its hybrid zone with the hooded crow. Mol Ecol. 2010, 19: 162-175.
Li W, Godzik A: Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006, 22 (13): 1658-1659.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009, 25: 1754-1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079.
Alexa A, Rahnenfuhrer J: topGO: Enrichment analysis for Gene Ontology. R Package Version. 2010, 2: 12.0-
Smit AFA, Hubley G, Green P: RepeatMasker Open-3.0. 1996–2010. http://www.repeatmasker.org
Iseli C, Jongeneel CV, Bucher P: ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999, 1999: 138-148.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliams H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948.
Yang Z: PAML 4:Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007, 24 (8): 1586-1591.
Raineri E, Ferretti L, Esteve-Codina A, Nevado B, Heath S, Pérez-Enciso M: SNP calling by sequencing pooled sample. BMC Bioinform. 2012, 13: 239-
Philipp EER, Kraemer L, Melzner F, Poustka AJ, Thieme S, Findeisen U, Schreiber S, Rosenstiel P: Massively Parallel RNA Sequencing Identifies a Complex Immune Gene Repertoire in the lophotrochozoan Mytilus edulis. PLoS One. 2012, 7 (3): e33091-doi:10.1371/journal.pone.0033091
Bettencourt R, Pinheiro M, Egas C, Gomes P, Afonso M, Shank T, Santos RS: High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics. 2010, 11: 559-
Bai Z, Zheng H, Lin J, Wang G, Li J: Comparative Analysis of the Transcriptome in Tissues Secreting Purple and White Nacre in the Pearl Mussel Hyriopsis cumingii. PLoS One. 2013, 8 (1): e53617-doi:10.1371/journal.pone.0053617
Huan P, Wang H, Liu B: Transcriptomic analysis of the clam Meretrix meretrix on different larval stages. Mar Biotechnol. 2012, 14: 69-78.
Hou R, Bao Z, Wang S, Su H, Li Y, Du H, Hu J, Wang S, Hu X: Transcriptome Sequencing and De Novo Analysis for Yesso Scallop (Patinopecten yessoensis) Using 454 GS FLX. PLoS One. 2011, 6 (6): e21560-doi:10.1371/journal.pone.0021560
Milan M, Coppe A, Reinhardt R, Cancela LM, Leite RB, Saavedra C, Ciofi C, Chelazzi G, Patarnello T, Bortoluzzi S, Bargelloni L: Transcriptome sequencing and microarray development for the Manila clam, Ruditapes philippinarum: genomic tools for environmental monitoring. BMC Genomics. 2011, 12: 234-
Coppe A, Bortoluzzi S, Murari G, Marino IAM, Zane L, Papetti C: Sequencing and Characterization of Striped Venus Transcriptome Expand Resources for Clam Fishery Genetics. PLoS One. 2012, 7 (9): e44185-doi:10.1371/journal.pone.0044185
Clark MS, Thorne MAS, Vieira FA, Cardoso JCR, Power DM, Peck LS: Insights into shell deposition in the Antarctic bivalve Laternula elliptica: gene discovery in the mantle transcriptome using 454 pyrosequencing. BMC Genomics. 2010, 11: 362-
Qin J, Huang Z, Chen J, Zou Q, You W, Ke C: Sequencing and de novo Analysis of Crassostrea angulata (Fujian Oyster) from 8 Different Develo** Phases Using 454 GSFlx. PLoS One. 2012, 7 (8): e43653-doi:10.1371/journal.pone.0043653
Shi Y, Yu C, Gu Z, Zhan X, Wang Y, Wang A: Characterization of the Pearl Oyster (Pinctada martensii) mantle transcriptome unravels biomineralization genes. Mar Biotechnol. 2013, 15: 175-189.
Joubert C, Piquemal D, Marie B, Manchon L, Pierrat F, Zanella-Cléon I, Cochennec-Laureau N, Gueguen Y, Montagnani C: Transcriptome and proteome analysis of Pinctada margaritifera calcifying mantal and shell: focus on biomineralization. BMC Genomics. 2010, 11: 613-
Eierman LE, Hare MP: Crassostrea virginica transcriptome. FigShare. 2013b, Available online at http://dx.doi.org/10.6084/m9.figshare.873865
Cheung F, Haas BJ, Goldberg SMD, May GD, **ao Y, Town CD: Sequencing Medicago truncatula expressed sequence tags using 454 Life Science technology. BMC Genomics. 2006, 7: 272-
Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH: Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008, 17 (7): 1636-1647.
Calvo GW, Luckenback MW, Allen SK, Burreson EM: Comparative field study of Crassostrea gigas and Crassostrea virginica in relation to salinity in Virginia. J Shellfish Res. 1999, 18: 465-473.
Chu FE, La Peyre JF, Burreson CS: Perkinsus marinus infection and potential defense-related activities in eastern oysters, Crassostrea virginica: Salinity effects. J Invertebr Pathol. 1993, 62 (3): 226-232.
Quilang J, Wang S, Li P, Abernathy J, Peatman E, Wang Y, Wang L, Shi Y, Wallace R, Guo X, Liu Z: Generation and analysis of ESTs from the eastern oyster, Crassostrea virginica Gmelin and identification of microsatellite and SNP markers. BMS Genomics. 2007, 8: 157-
Zhang L, Guo X: Development and validation of single nucleotide polymorphism markers in the eastern oyster Crassostrea virginica Gmelin by mining ESTs and resequencing. Aquaculture. 2010, 302: 124-129.
Yu H, He Y, Wang X, Zhang Q, Bao Z, Guo X: Polymorphism in a serine protease inhibitor gene and its association with disease resistance in the eastern oyster (Crassostrea virginica Gmelin). Fish Shellfish Immunol. 2011, 30 (3): 757-762.
Vijay N, Poelstra JW, Künstner A, Wolf JBW: Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol Ecol. 2013, 22: 620-634.
Acknowledgements
We thank Dr. Qi Sun and colleagues at Life Sciences Core Laboratories Computational Biology Service Unit (CBSU) of Cornell University for bioinformatics consultation. We are indebted to Dr. Suzy Strickler for a custom pipeline for dN/dS calculation. Thanks to Dr. **ming Guo for generously providing C. gigas genomic resources. We also thank Dr. Colleen Burge and members of the Hare Lab for comments on this manuscript. We also express gratitude to Sigma ** Grant-in-Aid of Research and Cornell University for funding.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Both authors conceived of and designed the study. LE collected field samples and water quality data, extracted RNA, performed the assembly, annotation and analysis of the transcriptome sequences, and drafted the manuscript. MH advised in the assembly, annotation and analysis of the transcriptome sequences, and was involved in the development of the manuscript. Both authors edited the manuscript and approve of the final paper.
Electronic supplementary material
12864_2013_6237_MOESM1_ESM.xlsx
Additional file 1: Table S1: Annotation of osmoregulatory candidate transcriptome reftigs from C. virginica. Each row is an osmoregulatory candidate reftig identified from the C. virginica transcriptome. The identification as an osmoregulatory candidate is from the match between the GenBank nr description (column 3) of the annotated reftig to the description of an osmoregulatory candidate identified by Zhang et al. [17] in the C. gigas genome. Information provided in the table for each reftig are the reftig length, GenBank nr description, nr e-value, UniProt match, UniProt e-value, UniProt ID, KEGG ID, and nucleotide sequence. (XLSX 2 MB)
12864_2013_6237_MOESM2_ESM.docx
Additional file 2: Table S2: Significantly enriched gene ontologies for cellular components in osmoregulatory candidate genes. Osmoregulatory candidate genes were compared to the complete set of annotated genes, ordered by functional category. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. Indentations represent the ‘parent’:‘child’ tiered relationship of GO terms with deeper indentations representing more specific terminology relative to the boldface level-three ‘parent’ terms shown as enriched in Figure 5. Table S3. Significantly enriched gene ontologies for molecular functions in osmoregulatory candidate genes. Osmoregulatory candidate genes were compared to the complete set of annotated genes, ordered by functional category. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. Indentations represent the ‘parent’:‘child’ tiered relationship of GO terms with deeper indentations representing more specific terminology relative to the boldface level-three ‘parent’ terms shown as enriched in Figure 6. Table S4. Significantly enriched gene ontologies for cellular components from the low salinity population. Significantly enriched gene ontologies in 1:0 asymmetric genes from the low salinity population are ordered by p-value. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. Table S5. Significantly enriched gene ontologies for cellular components from the high salinity population. Significantly enriched gene ontologies in 1:0 asymmetric genes from the high salinity population are ordered by p-value. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. Table S6. Significantly enriched gene ontologies for molecular function from the low salinity population. Significantly enriched gene ontologies in 1:0 asymmetric genes from the low salinity population are ordered by p-value. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. Table S7. Significantly enriched gene ontologies for molecular function from the high salinity population. Significantly enriched gene ontologies in 1:0 asymmetric genes from the high salinity population are ordered by p-value. The p-value is derived from a Fisher’s exact test implemented in topGO from Bioconductor. (DOCX 52 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Eierman, L.E., Hare, M.P. Transcriptomic analysis of candidate osmoregulatory genes in the eastern oyster Crassostrea virginica. BMC Genomics 15, 503 (2014). https://doi.org/10.1186/1471-2164-15-503
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-503