
Abstract
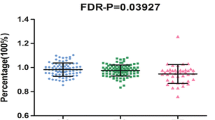
The Clusterin (CLU) gene, also known as apolipoprotein J (ApoJ), is currently the third most associated late-onset Alzheimer’s disease (LOAD) risk gene. However, little was known about the possible effect of CLU genetic variants on AD pathology in brain. Here, we evaluated the interaction between 7 CLU SNPs (covering 95% of genetic variations) and the role of CLU in β-amyloid (Aβ) deposition, AD-related structure atrophy, abnormal glucose metabolism on neuroimaging and CSF markers to clarify the possible approach by that CLU impacts AD. Finally, four loci (rs11136000, rs1532278, rs2279590, rs7982) showed significant associations with the Aβ deposition at the baseline level while genotypes of rs9331888 (P = 0.042) increased Aβ deposition. Besides, rs9331888 was significantly associated with baseline volume of left hippocampus (P = 0.014). We then further validated the association with Aβ deposition in the AD, mild cognitive impairment (MCI), normal control (NC) sub-groups. The results in sub-groups confirmed the association between CLU genotypes and Aβ deposition further. Our findings revealed that CLU genotypes could probably modulate the cerebral the Aβ loads on imaging and volume of hippocampus. These findings raise the possibility that the biological effects of CLU may be relatively confined to neuroimaging trait and hence may offer clues to AD.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common form of dementia in the elderly, accounting for 50% of all dementia1. It has been documented that genetic factors, along with environments, extremely contributes to the pathogenesis of AD2,3. Clusterin gene (CLU), also known as apolipoprotein J (ApoJ), is currently the third most associated risk gene according to Alzgene database (http://www.alzgene.org/). It is located in chromosome 8p21–p12 which is a chromosomal region of interest in AD4 and it may explain around 9% of the late-onset AD (LOAD) attributable risk5,6. Many large genome-wide association studies (GWAS) have identified that rs2279590, rs11136000, rs9331888, rs7012010, rs7982 and rs1532278 in CLU was substantially associated with AD risk in individuals of Caucasian ancestry and other populations7,8,9,10,11. Several independent candidate gene studies have then replicated and confirmed these results in various Caucasian populations or other populations, although the strongest associated variant sometimes differed12,13,14,15,16,17,18,19,20,21,22,23. Our group previously reported that rs9331949 and rs9331888 variation in the CLU gene played significant role in sporadic LOAD in the Han Chinese population24,25,26,57. However, no evidence supported that CLU genotypes impact the Aβ burden or tau in CSF in our study. More evidence may be needed to explain the interactions between CLU and Aβ burden in CSF.
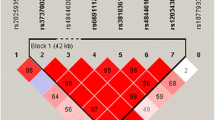
Genetically, multiple variations within CLU, such as rs2279590, rs11136000, rs9331888, rs7012010, rs9331949, rs7982 and rs1532278, have been identified to be associated with the risk of AD in multi-center, large scale GWAS, meta-analysis or replication studies. Among these loci, rs11136000 and rs9331888 were mostly investigated. Moreover, we performed linkage disequilibrium (LD) analysis and discovered that rs7982, rs11136000, rs1532278 and rs9331888 were in LD. The haplotypes (GCCG, ATTC) were related to the levels of amyloid deposition. Thus the haplotype-based analysis validated that CLU genotypes were related to the levels of amyloid deposition. The results presented here are not only correlative, but also support that CLU modulates the alteration of the biomarkers of Aβ markers to influence the risk of AD in vivo.
To date, continuous variable phenotypic analysis is now widely used to elucidate the specific role of genetics of multiple diseases. Distincted from the previous two categorical variable analysis (case vs control), the phenotypic analysis can not only be more sensitive to the association between genetic mutation and AD, but also provide more intuitively to explain the specific genetic effects on brain structure and function58. To date, numbers of GWAS–validated or GWAS-promising candidate loci have been certificated that they influence imaging and clinical features in AD40,59,60,61.
The advantage of our study is the method we use. Imaging genetics is an emergent transdisciplinary research field, in which genetic risk is assessed with imaging measures as quantitative traits (QTs) or continuous phenotypes. QT association studies have increased statistical power and decreased sample size requirements, thus imaging genetics studies have advantages over traditional case-control designs62,63. Although the differences across phenotypes with the same SNP might reflect power differences due to sample size differences, our findings that CLU modulates the alteration of the biomarkers of Aβ markers to influence the risk of AD in vivo were also supported that by animal studies from 10 years ago linking CLU/APOJ to amyloid deposition. Hence, the important role of this paper is that it confirmed the results of animal studies with in vivo neuroimaging data. However, the neuroimaging data were available only in a subset of participants in some QT analyses, e.g., half of participants with MRI information, 70% with FDG-PET, and 55% with AV45. Therefore, the QT analysis had a reduced sample size in some cases. Besides, the ADNI data was restricted to Caucasians to avoid genetics stratification across ethnicities. The 7 loci in CLU, however, have different frequencies in different races; therefore, our results cannot represent the other ethnicities, warranting the replications in other races.
In summary, our results showed that four loci (rs11136000, rs1532278, rs2279590, rs7982) showed significant associations with the Aβ deposition at the baseline level while genotypes of rs9331888 (P = 0.042) increased Aβ deposition. Besides, rs9331888 was significantly associated with baseline volume of left hippocampus (P = 0.014). We then further validated the association with Aβ deposition in the AD, mild cognitive impairment (MCI), normal control (NC) sub-groups. The results in sub-groups confirmed the association between CLU genotypes and Aβ deposition further. Moreover, our findings are also supported by animal studies from 10 years ago linking CLU/APOJ to amyloid deposition. These findings further supported the hypothesis that CLU genetic variations modulate the alteration of the biomarkers of Aβ markers to influence the risk of AD. These findings raise the possibility that the biological effects of CLU may be relatively confined to neuroimaging trait and hence may offer clues to the mechanisms through which particular genetic variants might influence AD risk.
Methods
ADNI dataset
The data in this study were obtained from Alzheimer’s Disease Neuroimaging Initiative (ADNI)64. ADNI is a large, multicenter, longitudinal neuroimaging study, launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and nonprofit organizations65. The initial goal of ADNI is to recruit 800 subjects. However, it has been followed by ADNI-GO and ADNI-2. Thus these three protocols have covered more than 1500 adults who are 55 to 90 years old to participate in the research, including cognitively normal (CN) older individuals, mild cognitive impairment (MCI), and early dementia patients with due to AD66. The study was approved by the institutional review boards of all participating centers (Ocean University of China, Qingdao Municipal Hospital, Nan**g First Hospital, Memory and Aging Center in University of California, and ADNI) and written informed consent was obtained from all participants or authorized representatives. In addition, the methods were carried out in accordance with the approved guidelines.
Participants
Participants were screened and enrolled according to criteria demonstrated in the ADNI study protocol (http://www.adni-info.org/scientists/adnistudyprocedures.aspx). We restricted the participants to whose genotype data of CLU SNPs were available and comprised 812 individuals. Baseline and longitudinal data including structural MRI and PET results were collected and all participants underwent a battery of clinical tests including Clinical Dementia Rating scale sum of boxes (CDRSB), Alzheimer’s disease Assessment Scale (ADAS-cog), Mini-Mental State Exam (MMSE), Rey Auditory Verbal Learning Test (RAVLT) and Functional Activities Questionnaire (FAQ) at baseline. According to the National Institute of Neurological and Communication Disorders/Alzheimer’s Disease and Related Disorders Association criteria for probable AD (NINCDS-ADRDA: probable AD), participants of AD were included if with a MMSE score between 20 and 26, a global Clinical Dementia Rating (CDR) of 0.5 or 1.0 and a CDRSB of 1.0 to 9.0. Amnestic MCI subjects achieved a MMSE score of 24 to 30 as well as a CDR score of 0.5 while the cognitively normal control individuals with a CDR score of 0. Furthermore, in this study, subjects with any serious neurological disease except for possible AD, any history of brain lesions or trauma, or psychoactive medication use (including antidepressants, neuroleptics, chronic anxiolytics, or sedative hypnotics) were excluded. In order to avoid population stratification effects which can lead to spurious genetic associations, we performed the principal component analysis (PCA). We assigned genotype-determined ancestry by comparing ADNI patients and populations form HapMap Phase 3 data and only individuals clustering with European HapMap samples were retained in our study.
SNP selection and Genoty**
Seven AD associated SNPs were selected for analysis.They have been validated to associate with AD in ethnically distinct populations7,8,9,10,11,21,28,67: rs2279590, rs11136000, rs9331888, rs7012010, rs9331949, rs7982, rs1532278. CLU genotypes were extracted from the ADNI GWAS PLINK format data68. We performed the quality control (QC) procedures using PLINK software. The inclusion criteria were as follows: minimum call rates >90%, minimum minor allele frequencies (MAF) > 0.01, Hardy-Weinberg equilibrium test P > 0.001.
PET measure-Aβ deposition
PET imaging data with amyloid tracer, florbetapir (AV-45), were obtained from UC Berkeley-AV45 analysis dataset on website (http://adni.loni.usc.edu/data-samples/access-data/). This institute used a native-space MRI scan for each subject which is segmented with Freesurfer (version 4.5.0) to define cortical grey matter regions of interest (ROI) (frontal, anterior/posterior cingulate, lateral parietal, lateral temporal) that make up a summary cortical ROI69,70. Notebly, the whole cerebellum was defined as reference region. Each florbetapir scan was applied to the corresponding MRI and mean florbetapir uptake within the cortical and reference region was calculated. Finally, SUVRs were created by averaging across the 4 cortical regions and dividing the cortical summary ROI by the whole cerebellum.
CSF Protein
CSF samples were collected and transported to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center in dry ice. Preparation of aliquots (0.5 ml) from the collected samples was conducted after thawing (1 h) at room temperature and gentle mixing. The aliquots were stored in bar code–labeled polypropylene vials at −80° C environment. The CSF proteins, including Aβ1-42, Total-tau and Phosphorylated tau181p, were calculated using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium; for research use-only reagents) immunoassay kit–based reagents. Additional analysis details and quality control procedures are showed at site (http://adni.loni.ucla.edu).The measurements of CSF biomarker for this article were cross-sectional from the baseline evaluation. Finally, a total of 501 individuals with genetic and other information were included in CSF analysis from the ADNI sites.
MRI structure
Our study used UCSF FreeSurfer datasets to conduct association test of CLU genotypes with brain structure. The cerebral image segmentation and analysis were performed with the FreeSurfer version 5.1 (http://surfer.nmr.mgh.harvard.edu/) based on the 2010 Desikan-Killany atlas71. We obtained data from motion correction and averaging of multiple volumetric T1 weighted images (when more than one is available), removal of non-brain tissue using a hybrid watershed/surface deformation procedure, automated Talairach transformation, segmentation of the subcortical white matter and deep gray matter volumetric structures (including hippocampus, amygdala, caudate, putamen, ventricles)72, intensity normalization, tessellation of the gray matter white matter boundary, automated topology correction, and surface deformation following intensity gradients to optimally place the gray/white as well as gray/cerebrospinal fluid borders at the location where the greatest shift in intensity defines the transition to the other tissue class. The technical details of these procedures are described in prior publications73.
PET measure-Glucose metabolism
FDG analysis data were from UC Berkeley and Lawrence Berkeley National Laboratory on the website (http://adni.loni.usc.edu/data-samples/access-data/)74. In this laboratory, five regions (left and right angular gyrus, bilateral posterior cingulate, left and right temporal gyrus) were treated as metaROIs (regions of interest) to analysis. Firstly, we downloaded the PET data from LONI (http://loni.usc.edu/). Then these images were spatially normalized in SPM to the MNI PET template. The mean counts from the metaROIs for each subject’s FDG scans at each time point were extracted and the intensity values were computed with SPM subroutines. Finally, the mean of the top 50% of voxels within a hand-drawn pons/cerebellar vermis region which was hand-drawn on a T1 template in MNI space was extracted. In addition, each metaROI mean was normalized by dividing it by pons/vermis reference region mean75.
Statistical Analysis
Differences in continuous variables were examined using one-way analysis of variance (ANOVA), and categorical data were tested using χ2 test. ADNI sample were stratified into three groups (CN, MCI and AD) to detect the effects of CLU genetic variations on neuroimaging phenotypes in the three clinical stages respectively. Moreover, we used a multiple linear regression model which considered age, gender, education, and ApoE ε4 status as covariates to estimate coefficients for testing possible correlation between various phenotypes and CLU genotypes. All statistical analyses were performed by R 3.12 and PLINK 8 (http://pngu.mgh.harvard.edu/wpurcell/plink/). To control multiple hypothesis testing, we used the false discovery rate (FDR) for correction76 and statistical significance was defined for FDR-corrected P < 0.05.
Additional Information
How to cite this article: Tan, L. et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci. Rep. 6, 26027; doi: 10.1038/srep26027 (2016).
References
Holtzman, D. M., Morris, J. C. & Goate, A. M. Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3, 77sr1 (2011).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63, 168–74 (2006).
Jiang, T., Yu, J. T., Tian, Y. & Tan, L. Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Curr Alzheimer Res 10, 852–67 (2013).
Wu, Z. C., Yu, J. T., Li, Y. & Tan, L. Clusterin in Alzheimer’s disease. Adv Clin Chem 56, 155–73 (2012).
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D. & Tanzi, R. E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39, 17–23 (2007).
Rajagopalan, P., Hibar, D. P. & Thompson, P. M. TREM2 and neurodegenerative disease. N Engl J Med 369, 1565–7 (2013).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41, 1088–93 (2009).
Lambert, J. C. et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41, 1094–9 (2009).
Seshadri, S. et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–40 (2010).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436–41 (2011).
Jun, G. et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21, 108–17 (2015).
Schjeide, B. M. et al. The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch Gen Psychiatry 68, 207–13 (2011).
Corneveaux, J. J. et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet 19, 3295–301 (2010).
Lee, J. H. et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol 68, 320–8 (2011).
Carrasquillo, M. M. et al. Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol 67, 961–4 (2010).
Kamboh, M. I. et al. Association of CLU and PICALM variants with Alzheimer’s disease. Neurobiol Aging 33, 518–21 (2012).
Liu, G. et al. The CLU gene rs11136000 variant is significantly associated with Alzheimer’s disease in Caucasian and Asian populations. Neuromolecular Med 16, 52–60 (2014).
Guerreiro, R. J. et al. Genetic variability in CLU and its association with Alzheimer’s disease. PLos one 5, e9510 (2010).
Klimkowicz-Mrowiec, A. et al. Lack of association of CR1, PICALM and CLU gene polymorphisms with Alzheimer disease in a Polish population. Neurol Neurochir Pol 47, 157–60 (2013).
Allen, M. et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 79, 221–8 (2012).
Chen, L. H. et al. Polymorphisms of CR1, CLU and PICALM confer susceptibility of Alzheimer’s disease in a southern Chinese population. Neurobiol Aging 33, 210 e1–7 (2012).
Jun, G. et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 67, 1473–84 (2010).
Roussotte, F. F., Gutman, B. A., Madsen, S. K., Colby, J. B. & Thompson, P. M. Combined effects of Alzheimer risk variants in the CLU and ApoE genes on ventricular expansion patterns in the elderly. J Neurosci 34, 6537–45 (2014).
Yu, J. T. et al. Implication of CLU gene polymorphisms in Chinese patients with Alzheimer’s disease. Clin Chim Acta 411, 1516–9 (2010).
Yu, J. T. et al. Genetic variation in clusterin gene and Alzheimer’s disease risk in Han Chinese. Neurobiol Aging 34, 1921 e17–23 (2013).
Tan, L. et al. Association of GWAS-linked loci with late-onset Alzheimer’s disease in a northern Han Chinese population. Alzheimers Dement 9, 546–53 (2013).
**ng, Y. Y. et al. Blood clusterin levels, rs9331888 polymorphism, and the risk of Alzheimer’s disease. J Alzheimers Dis 29, 515–9 (2012).
Yu, J. T. & Tan, L. The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy. Mol Neurobiol 45, 314–26 (2012).
Yu, J. T., Tan, L. & Hardy, J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci 37, 79–100 (2014).
Szymanski, M., Wang, R., Bassett, S. S. & Avramopoulos, D. Alzheimer’s risk variants in the clusterin gene are associated with alternative splicing. Transl Psychiatry 1, e18 (2011), doi: 10.1038/tp.2011.17.
Schurmann, B. et al. Association of the Alzheimer’s disease clusterin risk allele with plasma clusterin concentration. J Alzheimers Dis 25, 421–4 (2011).
Mullan, G. M. et al. Plasma clusterin levels and the rs11136000 genotype in individuals with mild cognitive impairment and Alzheimer’s disease. Curr Alzheimer Res 10, 973–8 (2013).
Elias-Sonnenschein, L. S. et al. Genetic loci associated with Alzheimer’s disease and cerebrospinal fluid biomarkers in a Finnish case-control cohort. PLos One 8, e59676 (2013).
Zhou, Y. et al. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLos One 9, e103187 (2014).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–6 (2002).
Toledo, J. B. et al. Nonlinear Association Between Cerebrospinal Fluid and Florbetapir F-18 beta-Amyloid Measures Across the Spectrum of Alzheimer Disease. JAMA Neurol 72, 571–81 (2015).
Grimmer, T. et al. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry 65, 927–34 (2009).
Jack, C. R., Jr. et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 257–62 (2011).
Peper, J. S., Brouwer, R. M., Boomsma, D. I., Kahn, R. S. & Hulshoff Pol, H. E. Genetic influences on human brain structure: a review of brain imaging studies in twins. Hum Brain Mapp 28, 464–73 (2007).
Biffi, A. et al. Genetic variation and neuroimaging measures in Alzheimer disease. Arch Neurol 67, 677–85 (2010).
Karas, G. B. et al. A comprehensive study of gray matter loss in patients with Alzheimer’s disease using optimized voxel-based morphometry. Neuroimage 18, 895–907 (2003).
Henneman, W. J. et al. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology 72, 999–1007 (2009).
Raji, C. A., Lopez, O. L., Kuller, L. H., Carmichael, O. T. & Becker, J. T. Age, Alzheimer disease, and brain structure. Neurology 73, 1899–905 (2009).
Giannakopoulos, P. et al. Possible neuroprotective role of clusterin in Alzheimer’s disease: a quantitative immunocytochemical study. Acta Neuropathol 95, 387–94 (1998).
Thambisetty, M. et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67, 739–48 (2010).
Yerbury, J. J. et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J 21, 2312–22 (2007).
Narayan, P. et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat Struct Mol Biol 19, 79–83 (2012).
DeMattos, R. B. et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 99, 10843–8 (2002).
Schrijvers, E. M., Koudstaal, P. J., Hofman, A. & Breteler, M. M. Plasma clusterin and the risk of Alzheimer disease. JAMA 305, 1322–6 (2011).
Nuutinen, T., Suuronen, T., Kauppinen, A. & Salminen, A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev 61, 89–104 (2009).
Sekar, S. et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging 36, 583–91 (2015).
Mulder, S. D., Nielsen, H. M., Blankenstein, M. A., Eikelenboom, P. & Veerhuis, R. Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro. Glia 62, 493–503 (2014).
Jack, C. R., Jr. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9, 119–28 (2010).
Mengel-From, J. et al. CLU genetic variants and cognitive decline among elderly and oldest old. PLos One 8, e79105 (2013).
Apostolova, L. G. et al. Brain amyloidosis ascertainment from cognitive, imaging, and peripheral blood protein measures. Neurology 84, 729–37 (2015).
Sihlbom, C., Davidsson, P., Sjogren, M., Wahlund, L. O. & Nilsson, C. L. Structural and quantitative comparison of cerebrospinal fluid glycoproteins in Alzheimer’s disease patients and healthy individuals. Neurochem Res 33, 1332–40 (2008).
Kauwe, J. S. et al. Fine map** of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer’s disease. PLos One 6, e15918 (2011).
Mattay, V. S., Goldberg, T. E., Sambataro, F. & Weinberger, D. R. Neurobiology of cognitive aging: insights from imaging genetics. Biol Psychol 79, 9–22 (2008).
Wang, H. F. et al. Effect of EPHA1 genetic variation on cerebrospinal fluid and neuroimaging biomarkers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. J Alzheimers Dis 44, 115–23 (2015).
Liu, Y. et al. Association between NME8 locus polymorphism and cognitive decline, cerebrospinal fluid and neuroimaging biomarkers in Alzheimer’s disease. PLos one 9, e114777 (2014).
Zhang, X. et al. Bridging Integrator 1 (BIN1) Genotype Effects on Working Memory, Hippocampal Volume, and Functional Connectivity in Young Healthy Individuals. Neuropsychopharmacology 40, 1794–803 (2015).
Meyer-Lindenberg, A. & Weinberger, D. R. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci 7, 818–27 (2006).
Potkin, S. G. et al. Genome-wide strategies for discovering genetic influences on cognition and cognitive disorders: methodological considerations. Cogn Neuropsychiatry 14, 391–418 (2009).
Weiner, M. W. et al. 2014 Update of the Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimers Dement 11, e1–e120 (2015).
Mueller, S. G. et al. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin N Am 15, 869–77, xi-xii (2005).
Petersen, R. C. et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–9 (2010).
Lin, Y. L. et al. Genetic polymorphisms of clusterin gene are associated with a decreased risk of Alzheimer’s disease. Eur J Epidemio 27, 73–75 (2012).
Saykin, A. J. et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement 6, 265–73 (2010).
Jagust, W. J. et al. Relationships between biomarkers in aging and dementia. Neurology 73, 1193–9 (2009).
Mormino, E. C. et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 132, 1310–23 (2009).
Desikan, R. S. et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–80 (2006).
Fischl, B. et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage 23 Suppl 1, S69–84 (2004).
Jack, C. R., Jr. et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 27, 685–91 (2008).
Landau, S. M. et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–8 (2010).
Landau, S. M. et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32, 1207–18 (2011).
Hochberg, Y. & Benjamini, Y. More powerful procedures for multiple significance testing. Stat Med 9, 811–8 (1990).
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; ; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. This work was also supported by grants from the National Natural Science Foundation of China (81471309, 81171209, 81371406).
Author information
Authors and Affiliations
Consortia
Contributions
J.T.Y. and L.T. design the whole study. L.T. analyzed the data, wrote the main manuscript text and prepared all figures. H.F.W collected the data from ADNI database and prepared the tables. M.S.T., C.C.T. and X.C.Z. helped analyze the data. D.M. and W.J.Y. helped collect the data from ADNI database. T.J. helped to revise the manuscript. All authors reviewed the manuscript. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tan, L., Wang, HF., Tan, MS. et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci Rep 6, 26027 (2016). https://doi.org/10.1038/srep26027
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26027
- Springer Nature Limited
This article is cited by
-
Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer’s disease neuropathology
Molecular Neurodegeneration (2021)
-
Informative combination of CLU rs11136000, serum HDL levels, diabetes, and age as a new piece of puzzle-picture of predictive medicine for cognitive disorders
Molecular Biology Reports (2019)
-
Association between clusterin concentration and dementia: a systematic review and meta-analysis
Metabolic Brain Disease (2019)
-
7B2 chaperone knockout in APP model mice results in reduced plaque burden
Scientific Reports (2018)
-
Correlation of rs9331888 polymorphism with Alzheimer’s disease among Caucasian and Chinese populations: a meta-analysis and systematic review
Metabolic Brain Disease (2017)