
Abstract
Evidence has shown that both sleep loss and daily caffeine intake can induce changes in grey matter (GM). Caffeine is frequently used to combat sleepiness and impaired performance caused by insufficient sleep. It is unclear (1) whether daily use of caffeine could prevent or exacerbate the GM alterations induced by 5-day sleep restriction (i.e. chronic sleep restriction, CSR), and (2) whether the potential impact on GM plasticity depends on individual differences in the availability of adenosine receptors, which are involved in mediating effects of caffeine on sleep and waking function. Thirty-six healthy adults participated in this double-blind, randomized, controlled study (age = 28.9 ± 5.2 y/; F:M = 15:21; habitual level of caffeine intake < 450 mg; 29 homozygous C/C allele carriers of rs5751876 of ADORA2A, an A2A adenosine receptor gene variant). Each participant underwent a 9-day laboratory visit consisting of one adaptation day, 2 baseline days (BL), 5-day sleep restriction (5 h time-in-bed), and a recovery day (REC) after an 8-h sleep opportunity. Nineteen participants received 300 mg caffeine in coffee through the 5 days of CSR (CAFF group), while 17 matched participants received decaffeinated coffee (DECAF group). We examined GM changes on the 2nd BL Day, 5th CSR Day, and REC Day using magnetic resonance imaging and voxel-based morphometry. Moreover, we used positron emission tomography with [18F]-CPFPX to quantify the baseline availability of A1 adenosine receptors (A1R) and its relation to the GM plasticity. The results from the voxel-wise multimodal whole-brain analysis on the Jacobian-modulated T1-weighted images controlled for variances of cerebral blood flow indicated a significant interaction effect between caffeine and CSR in four brain regions: (a) right temporal-occipital region, (b) right dorsomedial prefrontal cortex (DmPFC), (c) left dorsolateral prefrontal cortex (DLPFC), and (d) right thalamus. The post-hoc analyses on the signal intensity of these GM clusters indicated that, compared to BL, GM on the CSR day was increased in the DECAF group in all clusters but decreased in the thalamus, DmPFC, and DLPFC in the CAFF group. Furthermore, lower baseline subcortical A1R availability predicted a larger GM reduction in the CAFF group after CSR of all brain regions except for the thalamus. In conclusion, our data suggest an adaptive GM upregulation after 5-day CSR, while concomitant use of caffeine instead leads to a GM reduction. The lack of consistent association with individual A1R availability may suggest that CSR and caffeine affect thalamic GM plasticity predominantly by a different mechanism. Future studies on the role of adenosine A2A receptors in CSR-induced GM plasticity are warranted.
Similar content being viewed by others


Introduction
Caffeine is the most widely used psychoactive substance1. Given its efficacy in improving alertness2 and alleviating cognitive impairments caused by sleep deprivation3 or sleep restriction4, it is often consumed to combat drowsiness5. On the cerebral level, both acute sleep loss and daily caffeine intake can lead to a decrease in human grey matter (GM) volumes6,7,8,9,10 as measured by magnetic resonance imaging (MRI). It is unclear whether caffeine alleviates or exacerbates the GM changes induced by insufficient sleep.
Sleep deprivation11 and sleep restrictions12,13,14 impair brain structures and functionality in rodents. In humans, while the effects of chronic sleep restriction (CSR) on GM are unclear, inconsistent changes in brain structures have been observed following different sleep disruptions. One-night sleep restriction (i.e. measuring after a night of 3-h time in bed) led to cortical changes specifically in young age groups, including decreased GM in the thalamus, precuneus, and postcentral gyrus and increased GM in the insula6. However, multiple cortical and subcortical GM regions could also change in a non-linear fashion in a time course of 24- to 72 h during a total sleep deprivation7,8. Although it has been shown that one-month sleep restriction could lead to reduced white matter diffusivity15, GM plasticity after a longer sleep restriction remained to be investigated. On the other hand, chronic caffeine administration has been found to inhibit hippocampal neurogenesis and cell proliferation in adult rodents16,17,18, and daily caffeine intake was found to be associated with reduced cortical and hippocampal GM volume in humans9,10. We hypothesized that GM would be reduced after CSR, while a concomitant caffeine intake could exacerbate the CSR-induced GM reduction.
Effects of both caffeine and sleep loss are mediated in part by the adenosine system19,20. Partly serving as a byproduct of neuronal activity, the release of extracellular endogenous adenosine is increased throughout wakefulness21, and the binding of adenosine to the A1 adenosine receptor (A1R) leads to neural inhibition. Extended wakefulness, including partial or complete sleep deprivation can lead to increased levels of extracellular adenosine22 and upregulated A1R binding23. Caffeine, a nonselective adenosine receptor antagonist, enhances synaptic excitation24, thereby improving alertness19, enhancing vigilance2, and counteracting sleep the distributions of the genotype, together with age loss-induced cognitive decline3,4. The magnitude of cerebral and behavioral responses to caffeine and sleep loss can vary in association with the A1R availability25,26,27. Hence, we expect that one's baseline A1R binding potentials may also predict the magnitudes of GM plasticity induced by CSR and caffeine. In addition, both the sensitivity to caffeine effects on sleep28 as well as the distribution of A1R29 are known to be associated with the variant rs5751876 of the A2A adenosine receptor (A2AR) encoding gene, ADORA2A. Hence, we matched the participant groups by the distributions of the genotype, together with age, gender, body mass index, chrono type, and level of habitual caffeine intake4.
Since the vasoconstrictive effect of caffeine can render bias in morphology structure through altering perfusion30, we also quantified cerebral blood flow (CBF) as a covariate in the morphometry analysis. As an exploratory step, we also examined whether CBF was altered after chronic sleep restriction. Earlier studies reported inconsistent changes after different durations of sleep loss. One study reported a reduced absolute CBF (absCBF) in the attention network after acute sleep restriction (4 h in bed) in subjects with higher drowsiness but elevated absCBF in basal forebrain in those who remained alert31. Others studies reported reduced relative CBF (rCBF) in parahippocampus, fusiform, and prefrontal cortices32, while rCBF was increased in occipital cortices and insula33 after one-night sleep deprivation. A diffusion MRI study revealed that one month of sleep restriction (5.5 h in bed) could lead to a reduced diffusivity in the orbitofrontal gyri, superior occipital gyri, insula, and fusiform but increased diffusivity in supplementary motor area and cingulate gyrus15. How chronic sleep restriction affects CBF remains to be explored.
Methods
The study took place at the :envihab research facility of the German Aerospace Center (Cologne, Germany). The study protocol was approved by the Ethics Committee of the regional Medical Board (Ärztekammer Nordrhein) and the German Federal Office for Radiation Protection. The study execution followed the declaration of Helsinki, and all participants have provided their informed consent voluntarily in a written form. We registered the study on German Clinical Trial Registry DRKS (04.04.2018; detailed protocol see https://drks.de/search/en/trial/DRKS00014379). All the participants have given informed consent in written form. The recruitment and data collection took place from year 2018 to 2020.
Study procedure
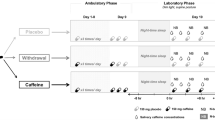
In a double-blind randomized study, 36 healthy adults (aged 28.9 ± 5.2 y/o; females: males = 15:21) underwent 9 consecutive laboratory days in the following order: 1-day adaptation (8-h time in bed), 2-day baseline (BL, 8-h time in bed/day), 5-day CSR days (5-h time in bed/day), 1-day recovery with 8 h time in bed (REC; Fig. 1). All participants are non-smokers with a habitual intake level of caffeine < 450 mg. Twenty-nine participants were homozygous C/C allele carriers of ADORA2A rs5751876, an A2AR gene variant, while 6 were heterozygous C/T allele carriers and 1 was homozygous T/T allele carriers. Inclusion/exclusion criteria and demographic data by groups are reported in Supplement (under Supplementary methods and Table S2) and Ref.27).

An overview of study protocol. The sleep restriction protocol consists of an adaptation day, two baseline days (BL), five sleep restriction days (CSR), and a recovery day (REC). The clock times of this protocol could be shifted according to the participant’s habitual bedtime, while the presented schedule is based on an 8 am–12 am wake-sleep schedule. On the nights of adaptation and the first BL days, participants had a full 8 h sleep (in blue shade). Starting from the second BL day throughout the first four CSR days, the bedtime was delayed by 3 h (i.e. 5-h time in bed). During the waking time (in while shade), participants were in constant luminance (~ 100 lx) during wakefulness. The coffee mug icons indicated the time when 19 participants received caffeine-containing coffee (CAFF group) and 17 received decaffeinated coffee (DECAF group). To monitor the caffeine levels, saliva samples were collected throughout the BL, CSR, and REC days (indicated by water drop icons). Three MRI scans took place in the afternoon of the second BL Day, the 5th CSR Day (average 7.0 ± 0.8 h after the last caffeine intake), and the REC Day (average 29.8 ± 4.3 h after the last caffeine intake), while for [18F]-CPFPX PET, we focused only on the baseline level measured on the second BL day.
All participants followed a one-week sleep satiation protocol (9 h in bed between 22:00 and 07:00 or between 23:00 and 08:00) prior to the start of the study. The compliance was monitored by daily self-reports and actimetry. Starting from the adaptation day, participants were in constant luminance (~ 100 lx) during wakefulness. Across the 5-day CSR, 19 of the 36 participants were assigned to the CAFF group and received daily coffee containing 200 mg (at 07:30 or 08:30) and 100 mg (at 14:00 or 15:00) of caffeine in the morning and at noon, respectively, while the other 17 participants assigned to the DECAF group received decaffeinated coffee4. [18F]-CPFPX PET-MRI acquisitions (Biograph mMR, Siemens) took place in the afternoon of the second BL Day, the 5th CSR Day (average 7.0 ± 0.8 h after the last caffeine intake), and the REC Day (average 29.8 ± 4.3 h after the last caffeine intake). To measure caffeine levels, salivary samples were collected repeatedly every evening (except for the adaptation day), as well as in the morning of the baseline days and before the PET scans (i.e. 1st and 2nd BL Day, 5th day of CSR and REC Day).
Randomization and intervention
We used the Visual Basic for Applications program of the Institute for medical biostatistics, informatics and epidemiology (IMBIE), University Bonn, for a stratified block randomization with the factors age, sex and BMI. For the caffeine administration for the CAFF group, we gave 200 mg caffeine in the morning and 100 mg caffeine in the afternoon in the form of coffee beverage. Meanwhile, DECAF group received identical volumes of decaf coffee on the same schedule. The procedure and device for the preparation of the caffeine-containing coffee and decaf coffee were standardized in accordance with a detailed instruction of the manufacturer, Tchibo GmbH, Coffee Technology, Hamburg, Germany. Using high-quality, electric drip filter coffee machines (Tchibo type 5794 and 2855) and a standardized brewing protocol, a consistent amount of caffeine was yielded: per 200 g regular coffee contains 101 ± 0.6 mg caffeine, while per 200 g decaf coffee contains 2.4 ± 0.05 mg caffeine (data available in Ref.4). Accordingly, both CAFF and DECAF group received 400 g coffee beverage in the morning and 200 g in the afternoon. All research team members involved in data collection and statistical analyses stayed blind to the randomization until the end of the study.
To confirm a successful caffeine administration, we monitored the salivary caffeine concentration daily throughout the laboratory protocol (Fig. 1). Saliva samples were taken in the mornings on the two BL Days, the 5th CSR Day, and REC Day as well as the evenings of all BL and CSR Days using Salivette® tubes (Sarstedt, Germany). Saliva samples were analyzed using an ultra-high-performance liquid chromatography system (Thermo Fisher, San Jose, CA), coupled with a linear ion trap quadrupole mass spectrometer 5500 (Sciex, Darmstadt, Germany; UHPLC-MS). For detailed protocol of sample preparation and UHPLC-MS analysis, please find the Method section of Ref.4.
Power calculations
Statistical power was calculated based on the primary endpoint of the parent project of this study. There, we hypothesized that chronic partial sleep deprivation would lead to lasting changes in neuroreceptors in the striatum of the brain. This increase in adenosine receptor availability, as compared the level after the 5-day chronic sleep deprivation to the baseline, was expected to be reduced by the regular consumption of caffeine compared to placebo. The adenosine receptor availability would be quantified by distribution volume (VT).
Accordingly, the number of subjects was calculated based on this hypothesis using an unpaired T-test. We based the expected effect (d = 0.91) on the results of our earlier study23 for the mean VT in the striatum (mean = 1.0 ml/ml). A statistical power of 0.8 was aimed for in order to find a significant mean difference of 0.1 ml/ml, with an assumed standard deviation of 0.11 ml/ml. Statistical significance level was set at p = 0.05. As a result, a sample size of 20 people per group was targeted.
Data acquisition and processing
After 3 dropouts in the DECAF group (85% completed) and 1 in the CAFF group (95% completed), we concluded the study with thirty-six subjects. Among the 36 subjects, one T1-weighted and Arterial Spin Labeling (ASL) images from the BL scan of a CAFF group subject were lost due to a technical reason on the scan day. In addition, during a CSR scan of another CAFF group subject as well as the REC scan of a DECAF group subject, the ASL images failed to be acquired.
In a series of stepwise analyses, we first identified the cortical changes in response to the main effect of CSR and the interaction between Caffeine and CSR. We conducted whole-brain multimodal voxel-wise morphometry (VBM) with linear mixed models, which allowed regressing out the variance of CBF voxel-to-voxel with ASL images. Next, we conducted a whole-brain exploratory analysis on ASL images to determine the main effect of CSR and the interaction effect between Caffeine and CSR on CBF. Lastly, we extracted the mean intensity of GM and mean CBF of the identified regions, as well as the binding potential (BPND) of A1R in cortical and subcortical regions from all time points (i.e. baseline, CSR Day, and REC Day), respectively. Using the extracted values, we then conducted post-hoc analyses as well as examined the association between the GM and/or CBF responses and the A1R BPND with linear regressions.
T1-weighted images and GM intensity
Grey matter (GM) morphology was assessed by T1-weighted images using 3-dimensional magnetization-prepared rapid acquisition with gradient echo (3D MPRAGE; isotropic voxel 1 mm, TR = 2250 ms, TE = 3.03 ms). For the preprocessing of T1-weighted images, we used the “Segment Longitudinal Data” pipeline provided by CAT12 toolbox on SPM12 (University College London, London, UK), which enabled a co-registration with the mean of the three volumes collected from the three timepoints (i.e. BL, CSR, and REC) of each participant and was suitable for repeated measurements. Affine registration was conducted using the tissue probability map in SPM12, followed by the segmentation of brain tissues into grey matter, white matter, cerebrospinal fluid, as well as total intracranial volume. The procedure continues with the Jacobian-modulated normalization using an MNI (Montreal Neurological Institute, McGill University)-defined standard brain. Lastly, we smoothed the preprocessed data with Gaussian kernel of FWHM = 8 × 8 × 8 mm3.
Next, we identified the GM responses to CSR and the interaction effect between caffeine and CSR using a voxel-wise multimodal analysis in order to statistically control for the potential bias caused by caffeine-induced changes in brain perfusion30,34,35. We used VoxelStats toolbox36, with which we conducted linear mixed models on two types of co-registered 3D volumes, i.e. T1-weighted and ASL, through each corresponding voxel. In addition to CBF, we used age, sex, and total intracranial volume as regressors of no interests in the model. The statistical significance of the coefficients was acquired with permutation tests (5000 times) and defined by a threshold at cluster-level pFDR < 0.001). To validate the novel toolbox VoxelStats and its voxel-to-voxel approach in tackling the bias of perfusion on morphometry, we compared the outcomes between controlling for voxel-wise CBF variances with VoxelStats and for global CBF variances with FMRIB Software Library (FSL 5.0; Oxford Center for Functional MRI of the Brain, United Kingdom) in the VBM analyses on T1-weighted images. Corresponding to the setup in Voxelstats, we used the “randomise” function (number of permutations = 5000) in FSL, and we used age, sex, and total intracranial volume as regressors as covariates. We present the statistical maps from VoxelStats and FSL in Supplementary method, Fig S1. In summary, the influence of CBF variances on the voxel and global level of the T1-weighted VBM analysis were strongly comparable. We believed that either (1) there was no significant contribution of CBF variances to the condition-driven GM changes (i.e. under caffeine or CSR) in general, or that (2) the within-subject comparison provided a good sensitivity to the condition-driven CBF variances and therefore well preserved the global variance to be reliably characterized on the voxel level. In any case, based on this precondition, we eventually decided to go for a voxel-wise approach as it could further take potential contributions of the regional CBF variances to the GM measurement into account, if any.
For the regions identified to be responsive to the interaction between caffeine and CSR, we further conducted a post-hoc analysis to determine the exact GM changes in each group in response to CSR using region-of-interest-based approach on R (R core team, Vienna, Austria). We first created masks of the responsive regions identified in the whole-brain analyses. Accordingly, we extracted mean signal intensity of these regions from all time points (BL, CSR, and REC) using FSL fslmeants, followed by a post-hoc analysis to determine the directions of GM responses in each group.
Cerebral blood flow
Cerebral blood flow (CBF) was measured by the sequence ASL (5 × 5 × 5 mm3, TR = 3500 ms, TE = 11 ms). For the preprocessing of ASL images, we used the standard pipeline on FSL. We used the first volume for the reference of motion correction as well as for the M0 calibration. We calculated the relative CBF maps with the acquired 40 tag-control pairs, followed by the quantification of absolute CBF using white matter as the reference tissue. Lastly, the absolute CBF maps were co-registered onto the T1-weighted images and MNI space.
Next, we conducted the whole-brain analysis on the absolute CBF maps with a GM mask using 2-way mixed effect ANOVA. Similar to the analysis on T1-weighted images, we also used nonparametric threshold-free cluster enhancement (number of permutations = 5000, cluster-level threshold pFDR < 0.01) with the “randomise” function. Same as the GM analysis, a post-hoc analysis was carried out on the significant changes in response to Caffeination x CSR using an identical procedure in extracting the regional mean response and post-hoc analyses.
Cerebral A1R availability
We acquired the [18F]-CPFPX PET data (framing scheme: 4 × 60 s, 3 × 120 s, 18 × 300 s, 2.09 × 2.09 × 2.03 mm3) simultaneously with the magnetic resonance imaging (MRI) scan. The scan in total lasted 100 min. We adopted an intravenous bolus injection simultaneously with the start of the scan, followed by a constant infusion (15.9 ml/ 34.1 ml, Kbol = 55 min.) The eventual dose injected was 175.9 ± 21.8 MBq and the molar activity was 102.25 ± 72.08 GBq/µmol at the time of injection.
For the preprocessing of PET, we used the standard pipeline of PMOD Neuro Tool (v4.006; PMOD Technologies). We first conducted motion correction with the average of the PET data collected in the first 9 min. The matching PET image was co-registered with the corresponding preprocessed T1-weighted image and segmented into grey matter, white matter, and cerebrospinal fluid, followed by the spatial normalization based on the MNI space. Seventy volumes of interests (VOIs) were defined by the automated anatomical labelling template of MNI space. For the kinetic modeling we used PMOD Kinetics Tool (version 4.006; PMOD Technologies). We examined the time-activity curves (TACs) and used cerebellum as a reference region to calculate the BPND of each volume of interest. The availability of A1R was indicated by the binding potential BPND of [18F]-CPFPX acquired with the Logan’s reference tissue model (t* = 30 min37).
We reported the details of procedure and parameters with regard to PET data acquisition, preprocessing, and kinetic modeling in the method section of Ref.27.
Predicting cerebral responses by cortical and subcortical A1R BPND
In a last step, we used A1R BPND to predict the GM responses identified. We first calculated the percent changes in CSR relative to BL using (meanCSR – meanBL)/meanBL × 100%. We calculated the A1R BPND data by cortical and subcortical regions. For cortical A1R BPND, we averaged the values acquired in frontal, occipital, parietal, temporal, and cingulate cortices, as well as amygdala, hippocampus, and insula. For subcortical A1R BPND, we averaged the values acquired in pallidum, olfactory tubercle, caudate nucleus, putamen, and thalamus by each hemisphere. The regions were segmented based on the automated anatomical labeling atlas (AAL38). We then conducted linear regression model to examine the coefficients for (1) caffeine effect, (2) interaction between caffeine effect and A1R BPND in predicting the percentage of the identified cerebral responses on R (R core team, Vienna, Austria). The A1R BPND used was taken from the hemisphere corresponding to the detected cerebral responses.
Results are presented as mean ± standard deviation.
Main results
Salivary concentration
On CSR Day 5, the CAFF group had a significantly higher salivary caffeine concentration than the DECAF group (CAFF: 2.24 ± 1.16 mg/L, DECAF: 0.08 ± 0.07 mg/L, tCAFF-DECAF = 9.9, p < 0.001). On REC Day, the salivary caffeine concentration in the CAFF group was lower (0.19 ± 0.31 mg/L, tCSR-REC = 9.7, p < 0.001) and reached a level, which did not significantly differ from the DECAF group (0.01 ± 0.02 mg/L, tCAFF-DECAF = 0.8, p = 0.871).
Multimodal analyses on grey matter
The results of whole-brain analyses are illustrated in Fig. 2, and the t values, p values, and Cohen’s d are detailed in Table 1. While no main effect of CSR on GM was found, the voxel-wise multimodal analysis controlled for the variances of CBF identified 7 large GM clusters where a significant interaction effect between caffeine and CSR was observed. The regions included temporal-occipital region: Cluster A1: right Rolandic operculum, Cluster A2: right middle occipital gyrus; dorsomedial prefrontal cortex (DmPFC): Cluster B1: bilateral medial superior frontal gyri and anterior cingulate cortex, Cluster B2: left sensory motor area and bilateral middle cingulate cortex; dorsolateral prefrontal cortex (DLPFC): Cluster C1: left middle frontal gyrus, Cluster C2: left precentral gyrus, inferior frontal operculum and insula; and Cluster D: right thalamus.

Regions showing significant differences between DECAF and CAFF groups in grey matter changes after CSR. The seven box plot panels display the clusters identified for a significant interaction effect between caffeine and CSR. The asterisks indicate a significant change compared to baseline as analyzed in post-hoc analyses. The color codes of the panels refer to a broader group of clusters based on functionality. The brain render visualizes the identified regions coded with the corresponding cluster number. The color bar indicates the t value acquired from the whole brain analysis. Finally, the render in multi-slices on the sagittal plane displays the exact location of the deeply seated Cluster D.
Interestingly, the post-hoc analysis revealed similarities among the different clusters in their response to CSR and recovery sleep. On CSR Day 5 compared to BL, the DECAF group exhibited a GM increase in all clusters. The CAFF group, however, showed a GM reduction in all clusters except right temporal-occipital cortex (Cluster A). On REC Day, the higher GM in the DECAF group had been remitted to the level of BL in all clusters except for left dorsolateral prefrontal cortex (Cluster C), which in general crosses the bilateral anterior and middle cingulate cortices. Furthermore, the GM reduction in the CAFF group remitted in all clusters except for thalamus (Cluster D). The response of thalamic GM inspired a supplementary correlation analysis between all the responsive regions (Supplements Fig S2), on which the following A1R availability analysis (see paragraph A1R BPND and GM plasticity) was based.
A1R BPND and grey matter plasticity
In order to reduce the bias from multiple comparisons, we synthesized GM responses into two groups – Cluster D (thalamus) and all others – based on the intercorrelation test between the Clusters responses (Supplement Fig S2). Overall, both cortical and subcortical A1R BPND could partly explain the group effect on the caffeine-associated GM reduction after CSR. However, subcortical A1R BPND showed a stronger and significant association with the caffeine-associated GM reduction after CSR, compared to cortical A1R BPND. Furthermore, neither cortical nor subcortical A1R BPND showed significant association with the GM changes in Cluster D (thalamus). The associations of cerebral responses and A1R BPND are presented in Fig. 3 with the corresponding stats in Table 2.

Associations between GM changes (% to baseline) and cortical/subcortical A1R BPND. (A) and (B) Display the associations of cortical and subcortical A1R BPND with GM responses, respectively. The statistical parameters are derived from analyses with linear mixed models. GM changes in all regions except thalamus are in blue while changes in thalamus are in red. Dark shades represent CAFF group while lighter shades represent DECAF group. The solid line indicates a significant association between baseline A1R availability and the GM change, while a dash line indicates an association at trend. Find the detailed statistics from the corresponding regression analyses in Table 2.
Exploratory analysis on cerebral blood flow
The whole-brain analysis on CBF did not indicate a significant difference between CSR Day 5 and BL Day or an interaction of caffeine with condition (i.e. CSR vs. BL)-BL. However, based on the solid evidence on the effects of caffeine and caffeine cessation on reducing and elevating CBF, respectively9,39,40,41,42,43,44,45,46, we examined the GM changes from CSR to REC, as well as a group difference in such changes. Interestingly, we found a significantly lower CBF on CSR compared to REC Day in the cerebellum, pons, brain stem, thalamus, hypothalamus, inferior temporal gyrus, and prefrontal cortex (all regions: pFDR-corrected < 0.05; Supplements Fig S3); however, we did not find a significant group difference in the divergent responses between CSR and REC. To provide further information, we conducted a series of supplementary analyses on the effect of CSR or REC compared to BL Day with a lower statistical threshold specifically in the regions showing lower CBF on CSR Day than REC Day (i.e. the medial frontal cortex, subcortical regions, occipital cortex, cerebellum, and midbrain). We found a reduced CBF on the CSR Day and an elevated CBF on REC Day compared to the BL Day in the CAFF group. The statistics and figures are presented in the Supplements Fig S3.
Discussion
The current study examined the cortical plasticity in response to CSR with or without the concomitant interference of caffeine. Independent of the caffeine-induced variances in CBF, we found opposite GM responses between the caffeine and decaffeinated groups after CSR. Specifically, GM in the left DLPFC, as well as right DmPFC, temporal-occipital, and thalamic cortices were increased after 5-day CSR without caffeine; in contrast, concomitant caffeine intakes during CSR led to a decrease in prefrontal and thalamic cortices. All the cortical changes, except for the DLPFC increase in the DECAF group and the thalamic decrease in the CAFF group, recovered to the baseline level after an 8-h sleep opportunity (which included an approximately 35 h withdrawal from caffeine for the CAFF group). Most importantly, individuals with a lower availability of subcortical A1R at baseline showed a larger caffeine-associated GM decrease after CSR in all responsive regions except for thalamus. Taken together, our data reveal region-dependent GM plasticity after CSR, which diverges between the presence and absence of caffeine. Furthermore, individual trait in A1R availability may play a resilient role against the effects of caffeine on GM changes after CSR.
Implications on use-dependent cortical and synaptic plasticity associated with adenosine systems
GM plasticity and synaptic strengths
Brain structural alterations induced by sleep loss have been frequently reported. Extensive sleep loss such as 36-h to 72-h sleep deprivation7,8 as well as one month of CSR15 was found to lead to reduced GM or structural network. Moreover, 21-h extended wakefulness followed by 3-h time in bed resulted in both reduced GM in thalamus, precuneus, and precentral gyrus as well as increased GM in insula6. Our data showing increased GM in DLPFC, DmPFC, thalamus, and temporal-occipital regions after 5 days of CSR with 5-h time in bed daily seemingly conflicts to the GM reduction commonly observed. However, the inconsistency may lie in the subtle differences in the sleep protocols – specifically, the total days of sleep restriction and the different amounts of sleep as daily recovery process. The synaptic homeostasis hypothesis (SHY)47 elaborated how synaptic homeostasis is maintained in sleep–wake cycles. It is suggested that the synaptic strengths are increased and saturated throughout wakefulness and downregulated as a restorative process during sleep47. Extensive sleep loss, therefore, may instead lead to neuronal exhaustion and hence the commonly observed GM reduction. However, partial sleep (i.e. 5-h time in bed) may allow partial brain restoration, which enables the synaptic strengths to maintain its upregulated state without an exhaustion within limited days of sleep restriction, therefore an increased GM in the current study. An earlier study examining the trajectory of GM changes throughout a 36-h sleep deprivation found a non-linear course of responses7. They observed increased GM in striatum and cingulate cortices throughout 36-h sleep deprivation, while a regional GM reduction only started to show at 32 h onward in the thalamus, insula, and somatosensory association cortex7. Their findings support the adaptability of brain structure to a certain extent of sleep deprivation until exhaustion might occur. On the contrary, caffeine has been shown to reduce the synaptic long-term potentiation (LTP) in rodents48 and LTP-like cortical activity in humans49, which may counteract the sleep loss-induced brain plasticity. In other words, the saturation and suppression of synaptic strengths might underlie the CSR-associated GM increase and caffeine-associated GM decrease, respectively.
Adenosine system in energy use-dependent synaptic and cortical plasticity
GM plasticity may occur in response to an increased or decreased need for energy resources, for which adenosine is a critical index as a byproduct of energy usage20,21. A study found a higher GM volume in the morning compared to the evening50 and suggested that the time of day-dependent plasticity of GM may be an evolutionary adaption to the energy consumption of diurnality50. In sleep homeostasis process, the elevated and reduced adenosinergic activity throughout wakefulness and sleep, respectively, is believed to be a biological index of the accumulation and dissipation of homeostatic sleep pressure21. By blocking adenosine receptors, caffeine counteracts the increased sleepiness as one of the classic effects and switches on the “energy saving mode” by reducing cortical activity51, cerebral metabolic rate of glucose52, and cerebral metabolic rate of oxygen (CMRO2)53,54. Our data show that caffeine does not only counteract the CSR-associated GM increase but further downregulate cortical intensity potentially through hindering the adenosine-mediated energy consumption, which is in line with earlier evidence of a reduced or lower GM volume after 10-day 450 mg caffeine intake9 or in habitual high-dose caffeine consumers10 in various brain regions.
The association between striatal A1R availability and cortical reduction after CSR with caffeine implicates a role of the adenosine system in the CSR- and caffeine-associated GM plasticity. Specifically, a higher A1R availability seems to serve a function of resilience against caffeine-associated GM reduction. Such an A1R-dependent resilience has also been found to be against cognitive impairments induced by sleep deprivation55. Interestingly, despite the cortical and subcortical A1R availability being highly correlated, we observe a much stronger association of caffeine-related GM reduction with subcortical than cortical A1R availability. In striatum, A1Rs antagonistically interact with A2AR through a formation of G-protein coupled-receptor heterodimer as a “concentration-dependent switch”56 that modulates presynaptic glutamate release based on the extracellular adenosine levels. In a regular extent of wakefulness, the tonic neuro-inhibition maintained by A1R is strengthened as the waking state lasts longer, leading to a reduced striatal presynaptic glutamate release and neural firing57,58. This effect is likely to underlie the lower GM in the evening compared to mornings50. In relatively shorter (e.g. 12 h or 24 h) sleep deprivation7 and sleep restriction, adenosine may be rapidly elevated22, triggering the phasic excitatory adenosine A2A receptors, which in turns inhibits A1R as well as enhances glutamate release56,59,60 and neural plasticity24,61,62, there by potentially increasing GM as observed in the DECAF group. However, a prolonged neuroexcitation by an extensive sleep loss, e.g. a 36-h or 72-h deprivation or one-month sleep restriction, might turn into a detrimental hyperexcitability63 and lead to a decreased brain structure7,8,15. Caffeine, on the other hand, acutely enhances striatal glutamate release by its predominant antagonism at A1R64. However, daily or chronic caffeine administration diminishes the effect of A1R antagonism on increasing glutamate level65, which can lead to suppressing LTP and neuroplasticity48,49, thereby potentially a reduced GM as observed in the CAFF group9,10. Furthermore, through a gradually reduced affinity of A2AR to caffeine over daily or chronic intake56,60, a concomitant exposure to high extracellular adenosine level such during CSR allows the A2AR rebinding to the endogenous adenosine even with the presence of caffeine, thereby regaining the inhibition to A1R through the A1R-A2AR heterodimers in striatum56,60. The association between striatal A1R availability and caffeine-associated GM reduction after CSR further implicate a role of A1R in synaptic homeostasis and GM plasticity during caffeine intake and sleep loss.
Beyond the use-dependent perspective: unique GM response in thalamus
Interestingly, although thalamic GM shows a consistent CSR-associated GM increase and caffeine-associated GM decrease, its recovery seems to be much slower than other responsive regions. An earlier study following up the trajectory of medial temporal GM recovery after the cessation of 10-day 450 mg caffeine intake suggested a recovery time at minimum 36 h44. The current study using a lower caffeine dose with a shorter intake period found a shorter time needed for a full recovery for most of the responsive regions, except for thalamus. Furthermore, the GM reduction in thalamus is not directly associated with the individual A1R availability. An earlier study found regional differences in the change in adenosine signaling after sleep deprivation66. While adenosine was rapidly accumulated and dissipated in basal forebrain and cortices after sleep deprivation and during recovery sleep, in thalamus, together with midbrain regions, showed a continuous decrease in adenosine concentration after sleep deprivation and even a few hours into recovery sleep66. Such differences in adenosinergic activity between thalamus and other brain regions might underlie the unique responses of thalamus in our data. Since thalamus is one of the most critical regions in sleep–wake regulations67,68, and the structural or functional alterations of thalamus are often associated with insomnia69,70,71,72,73,74 or hypersomnia75, it may be impacted by the concomitant CSR and caffeine intake in a more critical manner compared to other cortical regions.
Limitations and summary
A few limitations in this study require careful interpretation and further investigation in the future. First, although we preselected participants based on the ADORA2A polymorphism, the lack of A2AR availability data as derived from PET precludes a broader understanding of the receptor’s role in sleep loss induced GM changes. This limitation warrants more adenosine and A2AR-focusing studies on the effects of CSR and daily caffeine intake to be conducted. Second, it might be argued that the estimation of GM by MRI T1-weighted image can be potentially impacted by caffeine-induced changes in perfusion30. However, our study included both structural imaging and CBF measurement, and we did not observe associations between the two, including the pattern of changes across regions. An earlier study further suggests that the covariance between T1-weighted measurement and CBF rather occurs on a global instead of regional level9. Lastly, the caffeine and decaffeinated treatments in the current study were delivered as identically brewed coffee only different in the caffeine content, which indeed guaranteed the precision of caffeine administration. However, with other ingredients contained in brewed coffee, the observed outcomes could not be fully attributed to the effects of caffeine alone, but it could also be an effect from the interaction between caffeine and other biochemicals. Nevertheless, using brewed coffee is preferred given that it provides a better generalizability to a real-life coffee intake.
In summary, this study revealed reversible cortical plasticity in frontal, temporal-occipital, and thalamic GM in response to CSR. This plastic response, however, can be suppressed or reversed by concomitant caffeine intake. The individual baseline subcortical A1R availability may play a role in the caffeine-associated GM response. More studies are warranted to further investigate the role of A2AR in these GM changes.
Data availability
All the data reported in this manuscript is available for research purposes upon request. Please contact d.elmenhorst@fz-juelich.de for the request.
References
Heckman, M. A., Weil, J. & Gonzalez de Mejia, E. Caffeine (1, 3, 7-trimethylxanthine) in foods: A comprehensive review on consumption, functionality, safety, and regulatory matters. J. Food Sci. 75(3), R77-87 (2010).
Einother, S. J. & Giesbrecht, T. Caffeine as an attention enhancer: Reviewing existing assumptions. Psychopharmacology 225(2), 251–274 (2013).
Hansen, D. A. et al. Randomized, double-blind, placebo-controlled, crossover study of the effects of repeated-dose caffeine on neurobehavioral performance during 48 h of total sleep deprivation. Psychopharmacology 236(4), 1313–1322 (2019).
Baur, D. M. et al. Coffee effectively attenuates impaired attention in ADORA2A C/C-allele carriers during chronic sleep restriction. Prog. Neuro-psychopharmacol. Biol. Psychiatry 109, 110232 (2020).
Mahoney, C. R. et al. Intake of caffeine from all sources and reasons for use by college students. Clin. Nutr. 38, 668–675 (2018).
Long, Z., Cheng, F. & Lei, X. Age effect on gray matter volume changes after sleep restriction. PLoS One 15(2), e0228473 (2020).
Dai, X. J. et al. Plasticity and susceptibility of brain morphometry alterations to insufficient sleep. Front. Psychiatry 9, 266 (2018).
Liu, C., Kong, X. Z., Liu, X., Zhou, R. & Wu, B. Long-term total sleep deprivation reduces thalamic gray matter volume in healthy men. Neuroreport 25(5), 320–323 (2014).
Lin, Y.-S. et al. Daily caffeine intake induces concentration-dependent medial temporal plasticity in humans: A multimodal double-blind randomized controlled trial. Cereb. Cortex 31(6), 3096–3106 (2021).
Pham, K. et al. High coffee consumption, brain volume and risk of dementia and stroke. Nutr. Neurosci. 50, 1–12 (2021).
Abushov, B. M. Morphofunctional analysis of the effects of total sleep deprivation on the CNS in rats. Neurosci. Behav. Physiol. 40(4), 403–409 (2010).
Zhao, H. et al. Frontal cortical mitochondrial dysfunction and mitochondria-related beta-amyloid accumulation by chronic sleep restriction in mice. Neuroreport 27(12), 916–922 (2016).
de Vivo, L. et al. Loss of sleep affects the ultrastructure of pyramidal neurons in the adolescent mouse frontal cortex. Sleep 39(4), 861–874 (2016).
Novati, A., Hulshof, H. J., Koolhaas, J. M., Lucassen, P. J. & Meerlo, P. Chronic sleep restriction causes a decrease in hippocampal volume in adolescent rats, which is not explained by changes in glucocorticoid levels or neurogenesis. Neuroscience. 190, 145–155 (2011).
Lee, M. H. et al. Impact of sleep restriction on the structural brain network. Neuroreport 27(18), 1299–1304 (2016).
Wentz, C. T. & Magavi, S. S. Caffeine alters proliferation of neuronal precursors in the adult hippocampus. Neuropharmacology 56(6–7), 994–1000 (2009).
Han, M. E. et al. Inhibitory effects of caffeine on hippocampal neurogenesis and function. Biochem. Biophys. Res. Commun. 356(4), 976–980 (2007).
Kochman, L. J., Fornal, C. A. & Jacobs, B. L. Suppression of hippocampal cell proliferation by short-term stimulant drug administration in adult rats. Eur. J. Neurosci. 29(11), 2157–2165 (2009).
Urry, E. & Landolt, H.-P. Adenosine, caffeine, and performance: From cognitive neuroscience of sleep to sleep pharmacogenetics. Curr. Top. Behav. Neurosci. 25, 331–366 (2015).
Reichert, C. F., Deboer, T. & Landolt, H. P. Adenosine, caffeine, and sleep-wake regulation: State of the science and perspectives. J. Sleep Res. 31(4), e13597 (2022).
Porkka-Heiskanen, T. & Kalinchuk, A. V. Adenosine, energy metabolism and sleep homeostasis. Sleep Med. Rev. 15(2), 123–135 (2011).
Huang, Z.-L., Zhang, Z. & Qu, W.-M. Roles of adenosine and its receptors in sleep-wake regulation. Int. Rev. Neurobiol. 119, 349–371 (2014).
Elmenhorst, D. et al. Sleep deprivation increases A1 adenosine receptor binding in the human brain: A positron emission tomography study. J. Neurosci. Off. J. Soc. Neurosci. 27(9), 2410–2415 (2007).
Lopes, J. P., Pliássova, A. & Cunha, R. A. The physiological effects of caffeine on synaptic transmission and plasticity in the mouse hippocampus selectively depend on adenosine A(1) and A(2A) receptors. Biochem. Pharmacol. 166, 313–321 (2019).
Rogers, P. J. et al. Association of the anxiogenic and alerting effects of caffeine with ADORA2A and ADORA1 polymorphisms and habitual level of caffeine consumption. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 35(9), 1973–1983 (2010).
Elmenhorst, D. et al. Recovery sleep after extended wakefulness restores elevated A1 adenosine receptor availability in the human brain. Proc. Natl. Acad. Sci. U. S. A. 114(16), 4243–4248 (2017).
Pierling, A. L. et al. Cerebral A(1) adenosine receptor availability in female and male participants and its relationship to sleep. NeuroImage 245, 118695 (2021).
Retey, J. V. et al. A genetic variation in the adenosine A2A receptor gene (ADORA2A) contributes to individual sensitivity to caffeine effects on sleep. Clin. Pharmacol. Ther. 81(5), 692–698 (2007).
Hohoff, C. et al. ADORA2A variation and adenosine A(1) receptor availability in the human brain with a focus on anxiety-related brain regions: Modulation by ADORA1 variation. Transl. Psychiatry 10(1), 406 (2020).
Ge, Q. et al. Short-term apparent brain tissue changes are contributed by cerebral blood flow alterations. PLoS One 12(8), e0182182 (2017).
Poudel, G. R., Innes, C. R. & Jones, R. D. Cerebral perfusion differences between drowsy and nondrowsy individuals after acute sleep restriction. Sleep 35(8), 1085–1096 (2012).
Zhou, F. et al. Regional cerebral hypoperfusion after acute sleep deprivation: A STROBE-compliant study of arterial spin labeling fMRI. Medicine 98(2), e14008 (2019).
Elvsåshagen, T. et al. Cerebral blood flow changes after a day of wake, sleep, and sleep deprivation. NeuroImage 186, 497–509 (2019).
Field, A. S., Laurienti, P. J., Yen, Y. F., Burdette, J. H. & Moody, D. M. Dietary caffeine consumption and withdrawal: Confounding variables in quantitative cerebral perfusion studies?. Radiology 227(1), 129–135 (2003).
Laurienti, P. J. et al. Relationship between caffeine-induced changes in resting cerebral perfusion and blood oxygenation level-dependent signal. AJNR Am. J. Neuroradiol. 24(8), 1607–1611 (2003).
Mathotaarachchi, S. et al. VoxelStats: A MATLAB package for multi-modal voxel-wise brain image analysis. Front. Neuroinform. 10, 20 (2016).
Logan, J. et al. Distribution volume ratios without blood sampling from graphical analysis of PET data. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 16(5), 834–840 (1996).
Tzourio-Mazoyer, N. et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 15(1), 273–289 (2002).
Addicott, M. A., Peiffer, A. M. & Laurienti, P. J. The effects of dietary caffeine use and abstention on blood oxygen level-dependent activation and cerebral blood flow. J. Caffeine Res. 2(1), 15–22 (2012).
Addicott, M. A. et al. The effect of daily caffeine use on cerebral blood flow: How much caffeine can we tolerate?. Hum. Brain Mapp. 30(10), 3102–3114 (2009).
Vidyasagar, R., Greyling, A., Draijer, R., Corfield, D. R. & Parkes, L. M. The effect of black tea and caffeine on regional cerebral blood flow measured with arterial spin labeling. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 33(6), 963–968 (2013).
Jones, H. E., Herning, R. I., Cadet, J. L. & Griffiths, R. R. Caffeine withdrawal increases cerebral blood flow velocity and alters quantitative electroencephalography (EEG) activity. Psychopharmacology 147(4), 371–377 (2000).
Mathew, R. J., Barr, D. L. & Weinman, M. L. Caffeine and cerebral blood flow. Br. J. Psychiatry J. Ment. Sci. 143, 604–608 (1983).
Lin, Y. S. et al. Time to Recover From Daily Caffeine Intake. Front. Nutr. 8, 787225 (2021).
Couturier, E. G., Laman, D. M., van Duijn, M. A. & van Duijn, H. Influence of caffeine and caffeine withdrawal on headache and cerebral blood flow velocities. Cephalalgia Int. J. Headache 17(3), 188–190 (1997).
Kennedy, D. O. & Haskell, C. F. Cerebral blood flow and behavioural effects of caffeine in habitual and non-habitual consumers of caffeine: A near infrared spectroscopy study. Biol. Psychol. 86(3), 298–306 (2011).
Tononi, G. & Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81(1), 12–34 (2014).
Blaise, J. H., Park, J. E., Bellas, N. J., Gitchell, T. M. & Phan, V. Caffeine consumption disrupts hippocampal long-term potentiation in freely behaving rats. Physiol. Rep. 6(5), e13632 (2018).
Hanajima, R. et al. Effect of caffeine on long-term potentiation-like effects induced by quadripulse transcranial magnetic stimulation. Exp. Brain Res. 237(3), 647–651 (2019).
Trefler, A. et al. Impact of time-of-day on brain morphometric measures derived from T1-weighted magnetic resonance imaging. NeuroImage 133, 41–52 (2016).
Lin, Y.-S. et al. Working memory performance after daily caffeine intake: Compromised performance and reduced hippocampal activity. bioRxiv 48, 277 (2021).
Park, C. A. et al. The effects of caffeine ingestion on cortical areas: Functional imaging study. Magn. Reson. Imaging 32(4), 366–371 (2014).
Merola, A. et al. Map** the pharmacological modulation of brain oxygen metabolism: The effects of caffeine on absolute CMRO(2) measured using dual calibrated fMRI. NeuroImage 155, 331–343 (2017).
Yang, R. et al. Studying cerebral hemodynamics and metabolism using simultaneous near-infrared spectroscopy and transcranial Doppler ultrasound: A hyperventilation and caffeine study. Physiol. Rep. 3(4), e12378 (2015).
Elmenhorst, D. et al. Recovery sleep after extended wakefulness restores elevated A(1) adenosine receptor availability in the human brain. Proc. Natl. Acad. Sci. U. S. A. 114(16), 4243–4248 (2017).
Ciruela, F. et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J. Neurosci. Off. J. Soc. Neurosci. 26(7), 2080–2087 (2006).
Huang, Z.-L., Urade, Y. & Hayaishi, O. The role of adenosine in the regulation of sleep. Curr. Top. Med chem. 11(8), 1047–1057 (2011).
Lovatt, D. et al. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc. Natl. Acad. Sci. U. S. A. 109(16), 6265–6270 (2012).
Rombo, D. M., Ribeiro, J. A. & Sebastiao, A. M. Hippocampal GABAergic transmission: a new target for adenosine control of excitability. J. Neurochem. 139(6), 1056–1070 (2016).
Ferre, S. et al. Adenosine A1–A2A receptor heteromers: New targets for caffeine in the brain. Front. Biosci. J. Virtual Lib. 13, 2391–2399 (2008).
Cunha, R. A. How does adenosine control neuronal dysfunction and neurodegeneration?. J. Neurochem. 139(6), 1019–1055 (2016).
Costenla, A. R. et al. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur. J. Neurosci. 34(1), 12–21 (2011).
Rombo, D. M. et al. Synaptic mechanisms of adenosine A2A receptor-mediated hyperexcitability in the hippocampus. Hippocampus 25(5), 566–580 (2015).
Quarta, D. et al. Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-D-aspartate receptor stimulation. J. Neurochem. 91(4), 873–880 (2004).
Quarta, D. et al. Opposite modulatory roles for adenosine A1 and A2A receptors on glutamate and dopamine release in the shell of the nucleus accumbens. Effects of chronic caffeine exposure. J. Neurochem. 88(5), 1151–8 (2004).
Porkka-Heiskanen, T., Strecker, R.E., McCarley, R.W. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 99(3), 507–517 (2000).
Yang, Y. C., Hu, C. C., Huang, C. S. & Chou, P. Y. Thalamic synaptic transmission of sensory information modulated by synergistic interaction of adenosine and serotonin. J. Neurochem. 128(6), 852–863 (2014).
Brown, R. E., Basheer, R., McKenna, J. T., Strecker, R. E. & McCarley, R. W. Control of sleep and wakefulness. Physiol. Rev. 92(3), 1087–1187 (2012).
Li, M. et al. Abnormalities of thalamus volume and resting state functional connectivity in primary insomnia patients. Brain Imaging Behav. 13(5), 1193–1201 (2019).
Grau-Rivera, O. et al. Association between insomnia and cognitive performance, gray matter volume, and white matter microstructure in cognitively unimpaired adults. Alzheimer’s Res. Ther. 12(1), 4 (2020).
Huang, G. et al. Altered thalamic functional connectivity and cerebral blood flow in insomnia disorder: A resting-state functional magnetic resonance imaging study. Clin. Imaging 88, 17–23 (2022).
Zou, G. et al. Altered thalamic connectivity in insomnia disorder during wakefulness and sleep. Hum. Brain Mapp. 42(1), 259–270 (2021).
Kim, N., Won, E., Cho, S. E., Kang, C. K. & Kang, S. G. Thalamocortical functional connectivity in patients with insomnia using resting-state fMRI. J. Psychiatry Neurosci. JPN 46(6), E639–E646 (2021).
Kang, J. M. K. et al. Low white-matter integrity between the left thalamus and inferior frontal gyrus in patients with insomnia disorder. J. Psychiatry Neurosci. JPN 43(6), 366–374 (2018).
Hansen, P. N. et al. Severe hypersomnia after unilateral infarction in the pulvinar nucleus-a case report. BMC Neurol. 20(1), 442 (2020).
Acknowledgements
We express our sincere appreciation to all our participants for their volunteering and cooperation. We also thank the friendly support of Prof. Christian Cajochen, Dr. Carolin Reichert, and colleagues from Centre for Chronobiology at UPK Basel, as well as Prof. Hsiao-Ying Wey and colleagues from Martinos Center at Mass General Hospital.
Funding
This work was supported by the Institute for Scientific Information on Coffee (ISIC), the Swiss National Science Foundation, the Clinical Research Priority Program Sleep & Health of the University of Zurich, the Aeronautics Program of the German Aerospace Center, and respective institutional funds from all contributing institutions.
Author information
Authors and Affiliations
Contributions
Y.-S. L.: data curation, formal analysis, visualization, manuscript original drafting. D. L. & A. F.: data curation, investigation, project administration, manuscript review & editing. D. M. B., C. C. & C. L.: data curation, manuscript review & editing. B. N.: methodology, software, manuscript review & editing. A. B., D. A., & H.-P. L.: conceptualization, funding acquisition, resources, methodology, supervision, manuscript review & editing. E.-M. E. & D. E.: conceptualization, investigation, funding acquisition, methodology, supervision, manuscript review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, YS., Lange, D., Baur, D. et al. Repeated caffeine intake suppresses cerebral grey matter responses to chronic sleep restriction in an A1 adenosine receptor-dependent manner: a double-blind randomized controlled study with PET-MRI. Sci Rep 14, 12724 (2024). https://doi.org/10.1038/s41598-024-61421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61421-8
- Springer Nature Limited