
Abstract
With the establishment of large biobanks, discovery of single nucleotide variants (SNVs, also known as single nucleotide polymorphisms (SNVs)) associated with various phenotypes has accelerated. An open question is whether genome-wide significant SNVs identified in earlier genome-wide association studies (GWAS) are replicated in later GWAS conducted in biobanks. To address this, we examined a publicly available GWAS database and identified two, independent GWAS on the same phenotype (an earlier, “discovery” GWAS and a later, “replication” GWAS done in the UK biobank). The analysis evaluated 136,318,924 SNVs (of which 6289 reached P < 5e−8 in the discovery GWAS) from 4,397,962 participants across nine phenotypes. The overall replication rate was 85.0%; although lower for binary than quantitative phenotypes (58.1% versus 94.8% respectively). There was a 18.0% decrease in SNV effect size for binary phenotypes, but a 12.0% increase for quantitative phenotypes. Using the discovery SNV effect size, phenotype trait (binary or quantitative), and discovery P value, we built and validated a model that predicted SNV replication with area under the Receiver Operator Curve = 0.90. While non-replication may reflect lack of power rather than genuine false-positives, these results provide insights about which discovered associations are likely to be replicated across subsequent GWAS.
Similar content being viewed by others

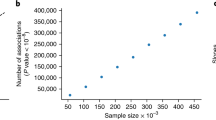

Introduction
Genome-wide association studies (GWAS) have resulted in the discovery of tens of thousands of genetic associations for various traits and phenotypes. Polygenic risk scores1, innovative drug discovery2, and gene-editing3 have all been enhanced, or even based on, GWAS results. Genome-wide association studies investigate the association of individual single nucleotide variants (SNVs) on a phenotype of interest (for example coronary artery diseases)4. Most GWAS identify SNVs with, individually, small effects4. This supports the notion that most diseases are polygenic, rather than monogenic, in nature5.
To observe the small effect of individual SNVs, GWAS have relied on increasingly larger sample sizes4. Recent advances have seen rapidly increasing sample sizes, particularly with the establishment of large biobanks. The most widely used and analyzed biobank in human genetics is the UK Biobank (UKBB)6. Analyses done in the UKBB and other similar biobanks have the opportunity not only to identify new associations but also to replicate previously proposed associations that arose from other GWAS investigations. It is not unexpected that some SNVs that were considered to be associated with a phenotype in an earlier GWAS may not be replicated in a subsequent GWAS. Even if they are replicated, their effect size may change, e.g. because of the winner’s curse phenomenon12,13,,13. With improved phenoty**, it seems plausible that these scores will continue to improve. Nevertheless, in the meantime there may be other ways to enhance current binary GWAS results for polygenic risk scores. First, our results clearly show a superior replication rate with quantitative phenotypes. These quantitative phenotypes are often more in line with physiological processes (e.g. systolic blood pressure) than clinical diseases (e.g. coronary artery disease). As such, future GWAS that directly use metabolomic data as outcomes (such as protein expression) are likely to, similarly, have higher accuracy than clinical disease phenotypes. Future research merging metabolomic outcomes and GWAS may be a useful addition to our scientific knowledge. For instance, some evidence suggests that the use of ‘intermediate’ phenotypes—between the genotype and the disease-based phenotype—may improve disease prediction14. For example, a 2021 study showed that the integration of polygenic risk scores for both disease-associated biomarkers and polygenic risk scores for the disease itself showed enhanced prediction over the polygenic risk score for the disease exclusively14. Second, almost all SNVs for binary traits with an OR > / = 1.2 were replicated, whereas the majority of SNVs with an OR below 1.2 were not replicated and this may reflect lack of power in the replication dataset. Of note, many of the replication UKBB datasets that we considered here did not use the full UKBB data, and power is likely to improve as complete biobank data are used and many biobanks are combined.
Limitations in comparison to previous literature
We were surprised to find only nine phenotypes where two GWAS had been conducted in truly independent participants and where inclusion or not of UKBB data was a distinguishing feature. It is plausible that further independent GWAS on the same traits exist, although this seems unlikely given the thorough and systematic search we performed of the GWAS atlas8. It is, however, likely that more GWAS are available, but they contain overlap** samples between GWAS (i.e. two GWAS of the same phenotype are not truly independent as they contain similar cohorts of participants), aren’t of sufficient quality to be included in the GWAS Atlas, are conducted in a non-European population, or have not made their summary statistics available. An earlier study15 reports building a model for SNV replication using GWAS for over 50 phenotypes, although it is unclear what, if any, measures were taken to determine if these numerous GWAS were truly independent i.e. did not include overlap** participants. Also, this study validated their model in two, small GWAS of one trait. Furthermore, this study didn’t actually quantify a SNV replication rate, nor did they stratify their results by binary and quantitative phenotypes. A further limitation of our study is that we didn’t include other SNV features, ideally we would have liked to include, for instance, LD as predictors in our model. However, this data was sparsely available. Lastly, it should be acknowledged that large disease-specific consortiums generally qualitatively describe the replication of SNVs as their consortium increases. Our study quantifies this formally and, importantly, quantifies replication across more than one phenotype.
Future research
We have identified a number of future research priorities. First, improving the phenoty** of binary phenotypes seems to be a priority for GWAS. Second, to facilitate an assessment of SNV replication, future independent cohorts are likely required. Many efforts to do this are already underway (e.g. AllofUs cohort and Millions Veteran Program).
Conclusions
The replication of SNVs discovered from GWAS was high for quantitative phenotypes. Genome-wide Association Studies appear to be entirely sufficient to detect SNVs associated with quantitative traits. For binary traits, however, the replication rate is modest. We have built a simple prediction model that can accurately ascertain SNV replication in later GWAS. It may be of use for researchers and clinicians that utilize GWAS results.
Data availability
All data used is publicly available from https://atlas.ctglab.nl/.
References
O’Sullivan, J. W. et al. Combining Clinical and Polygenic Risk Improves Stroke Prediction Among Individuals With Atrial Fibrillation. Circ Genom Precis Med. 14(3), e003168. https://doi.org/10.1161/CIRCGEN.120.003168 (2020).
Shu, L., Blencowe, M. & Yang, X. Translating GWAS findings to novel therapeutic targets for coronary artery disease. Front. Cardiovasc. Med. 5, 56 (2018).
Wu, S. et al. Genome-wide association studies and CRISPR/Cas9-mediated gene editing identify regulatory variants influencing eyebrow thickness in humans. PLoS Genet. 14, e1007640 (2018).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Lambert, S. A., Abraham, G. & Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 28, R133–R142 (2019).
Sudlow, C. et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
**ao, R. & Boehnke, M. Quantifying and correcting for the winner’s curse in genetic association studies. Genet. Epidemiol. 33, 453–462 (2009).
Watanabe, K. et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 51, 1339–1348 (2019).
Chinn, S. A simple method for converting an odds ratio to effect size for use in meta-analysis. Stat. Med. 19, 3127–3131 (2000).
Barendregt, J. J., Doi, S. A., Lee, Y. Y., Norman, R. E. & Vos, T. Meta-analysis of prevalence. J. Epidemiol. Commun. Health 67, 974–978 (2013).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults: Implications for primary prevention. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Abraham, G. et al. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat. Commun. 10, 5819 (2019).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 53, 185–194 (2021).
Gorlov, I. P. et al. SNP characteristics predict replication success in association studies. Hum. Genet. 133, 1477–1486 (2014).
Funding
There was no specific funding for this project. The lead author (JOS) was supported by an National Institutes of Health (NIH) T32 fellowship.
Author information
Authors and Affiliations
Contributions
J.O.S. and J.I. conceptualized the study design, J.O.S. attained and curated the data curation, J.O.S. performed the formal analysis; J.I. supervised the study; J.O.S. and J.I. drafted and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Sullivan, J.W., Ioannidis, J.P.A. Reproducibility in the UK biobank of genome-wide significant signals discovered in earlier genome-wide association studies. Sci Rep 11, 18625 (2021). https://doi.org/10.1038/s41598-021-97896-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97896-y
- Springer Nature Limited