
Abstract
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a new human disease with few effective treatments1. Convalescent plasma, donated by persons who have recovered from COVID-19, is the acellular component of blood that contains antibodies, including those that specifically recognize SARS-CoV-2. These antibodies, when transfused into patients infected with SARS-CoV-2, are thought to exert an antiviral effect, suppressing virus replication before patients have mounted their own humoral immune responses2,3. Virus-specific antibodies from recovered persons are often the first available therapy for an emerging infectious disease, a stopgap treatment while new antivirals and vaccines are being developed1,2. This retrospective, propensity score–matched case–control study assessed the effectiveness of convalescent plasma therapy in 39 patients with severe or life-threatening COVID-19 at The Mount Sinai Hospital in New York City. Oxygen requirements on day 14 after transfusion worsened in 17.9% of plasma recipients versus 28.2% of propensity score–matched controls who were hospitalized with COVID-19 (adjusted odds ratio (OR), 0.86; 95% confidence interval (CI), 0.75–0.98; chi-square test P value = 0.025). Survival also improved in plasma recipients (adjusted hazard ratio (HR), 0.34; 95% CI, 0.13–0.89; chi-square test P = 0.027). Convalescent plasma is potentially effective against COVID-19, but adequately powered, randomized controlled trials are needed.
Similar content being viewed by others

Main
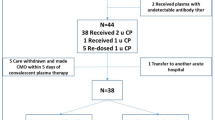
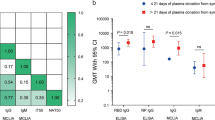
SARS-CoV-2 is a positive-sense, single-stranded RNA virus belonging to the family Coronaviridae. Humans infected with SARS-CoV-2 may develop COVID-19, which manifests across a wide spectrum of clinical severity ranging from a mild upper respiratory tract illness to a diffuse viral pneumonia causing acute respiratory failure, with sequelae including acute lung injury, multiorgan dysfunction syndrome and death4,5,6. Although protection from COVID-19 infection or disease has yet to be directly correlated with levels of circulating antibodies against SARS-CoV-2 (ref. 7), providing virus-neutralizing antibodies in the form of convalescent plasma may expedite disease resolution before the maturation of a patient’s own humoral response1, Both convalescent plasma treatment and retrospective analysis of data from our electronic medical record database were performed with the oversight of the Icahn School of Medicine at Mount Sinai (ISMMS) Institutional Review Board (IRB nos. 20-03574 and 20-03489). Convalescent plasma recipients were treated under compassionate use, via single-patient eIND applications to the FDA. As required by federal law, the ISMMS IRB was notified of every eIND application, FDA authorization was sought and received for each treated patient before transfusion, and all patients, or their legally authorized representatives, gave informed consent. As a retrospective analysis of compassionate-use treatment data, the study was neither prospectively designed nor registered on clinicaltrials.gov, nor was a data safety monitoring board prospectively convened to oversee this study. Between 24 March 2020 and 8 April 2020, 4,152 patients were hospitalized for COVID-19 in the MSHS. During this period, adult patients admitted to MSH were screened for eligibility to receive a COVID-19 convalescent plasma transfusion under the criteria established for the FDA single-patient eIND process, published 24 March 2020 (Supplementary Text 1). FDA eIND criteria included: age ≥18 years, severe or immediately life-threatening COVID-19 and patient or proxy ability to provide informed consent. Severe disease included at least one of the following: dyspnea; respiratory frequency ≥30 per min; blood oxygen saturation ≤93%; partial pressure of arterial oxygen to fraction of inspired oxygen ratio <300; and/or progression of lung infiltrates by >50% within 24–48 h. Life-threatening disease included at least one of the following: respiratory failure, septic shock and multiple-organ dysfunction or failure. There were no published exclusion criteria. As required by federal law (Code of Federal Regulations; 21CFR312.305 and 21CFR312.310), all patients treated under eIND criteria met the expanded access use requirements as documented on FDA form 3926, which was submitted for each individual patient and reviewed and approved by the FDA before transfusion. Initially, convalescent plasma inventory was limited relative to the number of eligible convalescent plasma recipients under eIND criteria; thus, patients were further prioritized by the following considerations, which were flexible according to plasma supply: (1) ABO blood type; (2) duration of symptoms; (3) length of stay, inclusive of admission at a transferring hospital; and (4) baseline functional status and comorbidities. Forty-five applications requesting individual patient eIND authorization to administer COVID-19 convalescent plasma were submitted to and approved by the FDA between 28 March and 8 April 2020. Sample size was not determined prospectively. The first eIND application was submitted upon receipt of the first convalescent plasma units from the New York Blood Center, and the last was submitted immediately before the MSHS joined the national Expanded Access Protocol (www.uscovidplasma.org/). Six patients consented to receive convalescent plasma and were granted FDA authorization under eIND criteria, but were not treated; four improved without convalescent plasma and two withdrew consent before transfusion. These six eINDs were withdrawn. Between the end of the study period (1 May 2020) and now (24 July 2020), two additional convalescent plasma recipients died of complications of multiorgan failure due to COVID-19, on days 42 and 88 after transfusion, for an overall death rate of 17.9%. The FDA was notified of all seven deaths and these eINDs were withdrawn. Thirty convalescent plasma patients were discharged from the hospital, for an overall discharge rate of 76.9%; these eINDs were withdrawn. One convalescent plasma recipient remains hospitalized as of 24 July 2020, and this eIND remains open. Convalescent plasma donors were screened for SARS-CoV-2 antibody titers with the MSH-ELISA anti-IgG COVID-19 assay, a two-step, spike-protein-directed ELISA23,24 adapted for emergency clinical use and performed by the Mount Sinai Laboratory25. Donors with total anti-spike IgG titers of ≥1:320 on the MSH-ELISA were referred for blood collection at the New York Blood Center, which performed the plasmapheresis and then returned convalescent plasma units and segments (apheresis tubing containing residual plasma) to MSH. Segments were frozen at −20 °C for research use. Convalescent plasma recipients were transfused with two units of ABO type-compatible convalescent plasma. The majority of recipients received both units from a single donor. Each unit, approximately 250 ml in volume, was infused over 1–2 h. Convalescent plasma recipients were monitored every 15 min for signs of transfusion-related reactions and then followed for outcomes after the transfusion. A propensity score–matched analysis was conducted within Epic electronic health records from the MSHS from 24 March 2020 to 8 April 2020. Analyses of patient data were performed using SAS 9.4. A logistic regression was fit to predict the potential for plasma therapy based on three sources of information: (1) baseline data, including age, gender, smoking status, obesity, diabetes, chronic obstructive pulmonary disease or sleep apnea and D-dimer and C-reactive protein at admission; (2) data from the day of transfusion (day 0), including supplemental oxygen requirement, length of hospital stay, minimal oxygen saturation, heart rate, respiratory rate and systolic and diastolic blood pressure; and (3) chronological data up to the day of transfusion, including the use of hydroxychloroquine or azithromycin, intubation status and, if intubated, the duration of intubation. Day 0 for convalescent plasma recipients was defined as the day on which they received plasma transfusion. For control patients, day 0 was defined as the day of hospitalization corresponding to the length of stay of their matched convalescent plasma recipient before transfusion. Two sets of matched data were generated based on 1:4 and 1:2 ratios for cases versus controls using the nearest neighbor matching algorithm, with and without replacement, respectively (SAS package: PROC PSMatch). Among the predictors, exact matching was enforced on oxygen requirement on the day of transfusion, length of hospital stay from the day of admission to the day of transfusion, the administration of hydroxychloroquine and azithromycin, and intubation status and duration (Supplementary Table 3). Other medications were administered too infrequently to enforce exact matching. The distribution of the logit propensity score–matched controls was within range of the convalescent plasma recipients, as opposed to the logit propensity score of the data from patients with confirmed COVID system wide (Extended Data Fig. 1). Balance was well achieved between the plasma and control groups, as all predictors had a standardized mean difference of less than 0.2 (Extended Data Fig. 2). Descriptive results for matched datasets are shown in Supplementary Table 3. Patients were evaluated for their supplemental oxygen requirements and survival at three time points: days 1, 7 and 14 after transfusion. Four categories of supplemental oxygen use status were collected for both cases and controls (Supplementary Tables 2 and 3). These included, in order of increasing severity: room air, without supplemental oxygen; low-flow oxygen delivery by standard nasal cannula; high-flow oxygen delivery, including non-rebreather mask, high-flow nasal cannula or BiPAP noninvasive ventilation; and mechanical ventilation. A patient’s oxygenation status at the three time points was considered to have worsened if they changed from a lower- to a higher-severity category compared to day 0, or if they had died before the time point. A generalized estimating equations approach with a logit link for binary data was used to model the effect of plasma on the odds of oxygenation improvement on days 1, 7 and 14 following transfusion, controlling for oxygen status on day 0. An independent working correlation structure was assumed for the patients within each cluster; however, P values were calculated based on the empirical standard errors. Since some patients with COVID-19 were being discharged with continued home oxygen supplementation during the study period, but this information was not easily obtainable from the database, the oxygen status of all discharged patients was assumed to be no worse than low-flow oxygen by standard nasal cannula. Adjusted covariates included duration of symptoms, use of pharmacotherapies (such as broad-spectrum antibiotics, therapeutic anticoagulation, azithromycin, corticosteroids, hydroxychloroquine and investigational antivirals) and laboratory values, specifically IL-6 levels. Kaplan–Meier survival curves were used to depict the overall survival following transfusion. A Cox model was fit to estimate the HR for in-hospital mortality for the plasma group, with matched clusters treated as random effects and onset of intubation as a time-varying covariate. In addition, interactions between convalescent plasma administration and intubation duration were tested to see if the plasma effects were the same in subgroups. Both survival models were adjusted for duration of symptoms before admission and other therapies administered during admission, as these data were only ascertained by manual chart review after the matching was completed. The initial list of therapies consisted of those used for COVID-19 during the study period, which included azithromycin, broad-spectrum antibiotics, hydroxychloroquine, therapeutic anticoagulants, corticosteroids and investigational therapies, including directly acting antivirals, mesenchymal stem cells and IL-1 and IL-6 inhibitors. Only those that had a chi-square test P value < 0.5, however, were included in the final model for adjustment. A liberal P value was used here to be inclusive of any potential confounders. As a sensitivity analysis, the 1:2 matching data without replacement data were also analyzed, where the balance between the matched pairs was enhanced but the study power was reduced. The MSH-ELISA anti-IgG COVID-19 assay23,24,25 is an orthogonal immune assay specific for anti-SARS-CoV-2 spike protein IgG in clinical serum or plasma specimens. It measures the relative concentration of IgG and reports the result as the reciprocal of the highest dilution of serum or plasma giving a positive signal. The assay received FDA emergency use authorization for clinical use on 15 April 2020 (https://www.fda.gov/media/137032/download/) and was also independently authorized as a laboratory developed test for clinical application by the New York State Department of Health at the Mount Sinai Laboratory, Center for Clinical Laboratories, a division of the Department of Pathology, Molecular and Cell-Based Medicine, New York (Clinical Laboratory Improvement Amendments no. 33D1051889). Plasma unit samples were retrieved from frozen apheresis segments, and antibody titers were determined by ELISA, performed in 96-well microtiter plates (Thermo Fisher) coated with 50 μl of recombinant full-length spike protein at a concentration of 2 μg ml−1 overnight at 4 °C. The next day, the plates were washed three times with PBS (Gibco) containing 0.1% Tween-20 (T-PBS; Fisher Scientific). The plates were blocked with 200 μl of blocking solution (T-PBS with 3% wt/vol milk powder (American Bio)) and incubated for at least 1 h at room temperature. Plasma samples were serially diluted in 1% milk prepared in T-PBS and added to the plates after the blocking solution was removed, and then the plates were incubated for 2 h at room temperature. The plates were washed three times with T-PBS using an automatic plate washer (BioTek) and 50 μl of anti-human IgG (Fab-specific) horseradish peroxidase antibody (produced in goat; Sigma, A0293), diluted 1:3,000 in T-PBS containing 1% milk powder, was added to all wells. After 1 h, the plates were washed three times with T-PBS, 100 μl of SigmaFast o-phenylenediamine dihydrochloride (Sigma) was added to all wells, and the reaction stopped after 10 min by adding 50 μl per well of 3 M hydrochloric acid (Thermo Fisher). The plates were read at a wavelength of 490 nm using a plate reader (BioTek), and the endpoint titer was calculated, defined as the last dilution before the signal dropped below an OD490 of 0.15. Donor serum samples were heat inactivated at 56 °C for 1 h before use. Vero.E6 cells from the American Type Culture Collection (ATCC no. CRL‐1586) were seeded at a density of 20,000 cells per well in a 96-well cell culture plate (Corning, 3595) 1 d before the assay was performed. Cells were maintained in culture in complete DMEM (Gibco), and the medium used for the neutralization assay was 1× MEM (Gibco) supplemented with 2% FBS (Corning). Starting with 1:10, serial dilutions of each sample in duplicate were prepared in a 96-well plate. Six wells in each plate were used as no-virus negative controls, and six wells were used as serum-free, virus-only positive controls. Next, 80 μl of each respective dilution was mixed with 600 median tissue culture doses (TCID50) of SARS‐CoV‐2 isolate USA‐WA1/2020 (BEI Resources, NR‐52281) in 80 μl. The virus–serum mixture was incubated for 1 h. The medium was removed from cells, and 120 μl of virus–serum mixture was added. After 1 h of incubation at 37 °C, the virus–serum mixture was removed, and 100 μl of MEM and 100 μl of each serum dilution was added to the cells. The cells were incubated at 37 °C for 2 d. The medium from the cells was removed, and 150 μl of 10% formaldehyde (Polysciences) was added for 24 h to fix cells and inactivate the virus. The next day, cells were permeabilized and stained using a mouse monoclonal anti-SARS-CoV-2 nucleocapsid antibody (clone 1C7, generated in-house by T. Moran, Center for Therapeutic Antibody Discovery at ISMMS). For each dilution, the inhibition of virus growth, relative to the controls, was calculated. A nonlinear regression was performed in Prism version 7.0 (GraphPad Software) to calculate the 50% inhibitory dilution (ID50), the serum dilution at which virus growth was halved relative to the serum-free, virus-only controls. The 50% neutralizing titer was defined as the reciprocal of the ID50. Donor serum antibody titers (total IgG against recombinant SARS-CoV-2 full-length spike protein, measured by the clinical MSH-ELISA) were available for all 25 donors who provided the convalescent plasma to this recipient cohort. Stored samples of donor serum, drawn on the same day as the antibody screening test, and unit plasma, retrieved from plasma segments, were available from 24 of 25 donors. The donor for whom these samples were not available had a screening antibody titer of 1:960 and donated both units of plasma to recipient number 15, who expired during the study period. The serum from one donor, who had a screening antibody titer of 1:320, showed no detectable neutralization of SARS-CoV-2 in the microneutralization assay; the 50% neutralization titer of this sample was set at 1 for the purposes of calculating geometric means and correlation coefficients. These data are provided as source data. For recipients who received plasma units from two different donors, the GM-NT of the serum samples from the two donors is presented in Fig. 3c. Spearman’s rank correlation coefficients (ρ) and GM-NTs were calculated in Prism version 8.0 (GraphPad). We did not perform a priori sample size calculations. The convalescent plasma-recipient cohort (n = 39) was a sample of convenience that included all adult patients treated with convalescent plasma under FDA eIND at MSH. The untreated control sample size was determined by matching at 1:2 and 1:4 ratios (cases to controls) by propensity score analysis. Group differences were evaluated by chi-square and Wilcoxon rank-sum tests for categorical and ordinal data, respectively. ORs and HRs are presented with 95% CIs and chi-square P values. Spearman’s rank correlation was used to test for monotonous relationships between plasma donors’ antibody titers and plasma recipients’ clinical outcomes, and analysis of variance was assessed by the Kruskal–Wallis test, followed by Dunn’s multiple-comparison test. All tests were two-sided, and statistical significance was defined as a P value < 0.05, unless otherwise indicated. Descriptive data are reported as number (percent), mean ± s.d., geometric mean (95% CI) or median (range or IQR), as appropriate. Further information on research design is available in the Nature Research Reporting Summary linked to this article.Methods
Ethics and regulatory oversight
Eligibility and selection of convalescent plasma recipients
Convalescent plasma transfusion
Propensity score matching of controls to convalescent plasma recipients
Assessment of respiratory status
Assessment of outcomes
Antibody assays and analyses
The MSH anti-IgG COVID-19 enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay
Microneutralization assay
Data analysis
Statistical analyses
Reporting Summary
Data availability
Data will be shared in accordance with ISMMS policy on access to and use and disclosure of Mount Sinai data. This process can be initiated upon request to the corresponding author. Source data are provided with this paper.
Code availability
No custom code is associated with this manuscript. The SAS code for these analyses can be downloaded from https://www.researchgate.net/project/Convalescent-plasma-treatment-of-severe-COVID-19-A-propensity-score-matched-control-study/.
References
Casadevall, A. & Pirofski, L. A. The convalescent sera option for containing COVID-19. J. Clin. Invest. 130, 1545–1548 (2020).
Rojas, M. et al. Convalescent plasma in COVID-19: possible mechanisms of action. Autoimmun. Rev. 19, 102554 (2020).
Sharun, K. et al. Antibody-based immunotherapeutics and use of convalescent plasma to counter COVID-19: advances and prospects. Expert Opin. Biol. Ther. 20, 1033–1046 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities and outcomes among 5,700 patients hospitalized with COVID-19 in the New York City area. JAMA 323, 2052–2059 (2020).
Duan, K. et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl Acad. Sci. USA 117, 9490–9496 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Zhao, J. et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa344 (2020).
Chen, L., **ong, J., Bao, L. & Shi, Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis. 20, 398–400 (2020).
Roback, J. D. & Guarner, J. Convalescent plasma to treat COVID-19: possibilities and challenges. JAMA 323, 1561–1562 (2020).
Luke, T. C., Kilbane, E. M., Jackson, J. L. & Hoffman, S. L. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann. Intern. Med. 145, 599–609 (2006).
Mair-Jenkins, J. et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J. Infect. Dis. 211, 80–90 (2015).
Enria, D. A., Briggiler, A. M., Fernandez, N. J., Levis, S. C. & Maiztegui, J. I. Importance of dose of neutralising antibodies in treatment of Argentine haemorrhagic fever with immune plasma. Lancet 2, 255–256 (1984).
Lee, J. S. et al. Anti-Ebola therapy for patients with Ebola virus disease: a systematic review. BMC Infect. Dis. 19, 376 (2019).
Ye, M. et al. Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. J. Med. Virol. https://doi.org/10.1002/jmv.25882 (2020).
Salazar, E. et al. Treatment of COVID-19 patients with convalescent plasma. Am. J. Pathol. 190, 1680–1690 (2020).
Perotti, C. et al. Mortality reduction in 46 severe COVID-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter interventional trial. Haematologica https://www.haematologica.org/content/early/2020/07/20/haematol.2020.261784.long (2020).
Gharbharan, A. et al. Convalescent plasma for COVID-19. A randomized clinical trial. Preprint at https://www.medrxiv.org/content/10.1101/2020.07.01.20139857v1 (2020).
Li, L. et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA 324, 460–470 (2020).
Finland, M. The serum treatment of lobar pneumonia. N. Engl. J. Med. 202, 1244–1247 (1930).
Cecil, R. L. Remarks on HE SERUM TREATMENT OF PNEUMONIA. Br. Med. J. 2, 657–662 (1932).
Hung, I. F. N. et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A (H1N1) infection. Chest 144, 464–473 (2013).
Austin, P. C. A comparison of 12 algorithms for matching on the propensity score. Stat. Med. 33, 1057–1069 (2014).
Amanat, F. et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 26, 1033–1036 (2020).
Stadlbauer, D. et al. SARS-CoV-2 seroconversion in humans: a detailed protocol for a serological assay, antigen production and test setup. Curr. Protoc. Microbiol. 57, e100 (2020).
Wajnberg, A. et al. SARS-CoV-2 infection induces robust, neutralizing antibody responses that are stable for at least three months. Preprint at http://medrxiv.org/content/early/2020/07/17/2020.07.14.20151126.abstract (2020).
Choe, P. G. et al. Antibody responses to SARS-CoV-2 at 8 weeks postinfection in asymptomatic patients. Emerg. Infect. Dis. 26 (2020).
Klein, S. et al. Sex, age and hospitalization drive antibody responses in a COVID-19 convalescent plasma donor population. J. Clin. Invest. https://doi.org/10.1172/JCI142004 (2020).
Okba, N. M. A. et al. Severe Acute Respiratory Syndrome Coronavirus 2-specific antibody responses in coronavirus disease patients. Emerg. Infect. Dis. 26, 1478–1488 (2020).
Joyner, M. J. et al. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin. Proc. https://www.mayoclinicproceedings.org/article/S0025-6196(20)30651-0/fulltext (2020).
Joyner, M. J. et al. Early safety indicators of COVID-19 convalescent plasma in 5,000 patients. J. Clin. Invest. 130, 4791–4797 (2020).
Llitjos, J. F. et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 18, 1743–1746 (2020).
Acknowledgements
This study was supported by internal funding from MSH and ISMMS. We thank all of the patients who participated in this study and their families. We also acknowledge the generosity of the thousands of anonymous tristate area residents who recovered from COVID-19 and then volunteered to donate convalescent plasma for the benefit of others. We thank the New York Blood Center, L.-A. Pirofski, T. Schneider, C. Seah, S. Srinivas, D. Tremblay, M. Geiger, C. Lebovits and J. Lustgarten. We acknowledge the assistance of ISMMS medical students: S. Ahsanuddin, A. Paasewe, R. Upadhyay, G. Mellgard, T. Martinson, B. Patil, C. Luo, S. Agrawal, A. Siddiqui, J. Schwarz, L. Piendel, J. Emerson, H. Kaplan, E. Klein, M. Garcia, J. Johnson, L. Maillie and E. Baldwin. We also appreciate the clinical expertise of the Mount Sinai Convalescent Plasma Squad: N. Shuman, D. Delbeau, D. Catamero, G. Sanchez, S. Aird, M. Mann, T. Broome, S. Kleiner-Arje, L. Wolf, A. Lee, L. Gaynes and K. Goodman. We dedicate this work to the New Yorkers who have lost their lives to COVID-19 with a special dedication to the health care workers who will always be remembered for their selflessness during this pandemic.
Author information
Authors and Affiliations
Contributions
J.A.A., A.C., N.M.B., S.T.H.L., H.-M.L., B.K.C. and D.L.R. conceived and designed the study. S.T.H.L., H.-M.L., P.T., M.A.L., E.B., A.D., C.S. and A.Z. analyzed the data. S.T.H.L., N.M.B., J.A.A., J.P.G., F.R., D.R.A., I.B., D.R.M., A.F.B., C.C.-C., J.S.J., S.A.A., A.W., J.B., D.R., A.B.-M. and F.K. participated in the donor plasma collection program and the transfusion of convalescent plasma recipients. Mount Sinai laboratory technicians performed the clinical MSH-ELISA assay, developed by F.A., D.S. and F.K. and transitioned to clinical use by D.R.M., A.F.-B. and C.C.-C. on donor serum samples. F.T.N., F.A. and D.S. performed the lab-based ELISA and microneutralization assays on donor serum and plasma samples, and N.M.B. analyzed the antibody titer data. S.T.H.L., H.-M.L. and N.M.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
F. Krammer has filed patent applications for the assay used to select plasma donors, and Mount Sinai has licensed its use to several companies. All other authors have nothing to disclose.
Additional information
Peer review information Alison Farrell is the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Propensity scoring of matched controls and all COVID-positive patients versus convalescent plasma recipients.
The logits of propensity scores were more similarly distributed for convalescent plasma recipients (n=39) and their propensity score-matched controls (n=156) than for all COVID-19 patients admitted to the Mount Sinai Health System between 24 March 2020 and 8 April 2020 (n=4,152). The means are represented by open circles (for convalescent plasma recipients) or plus signs (for matched controls and all COVID patients). The box represents the interquartile range, the center line is the median value, and the whiskers delineate the range between minimum and maximum values.
Extended Data Fig. 2 Standardized mean differences of matched controls and all COVID-positive patients.
Balance was well achieved between the groups of convalescent plasma recipients (n=39) and matched controls (n=156). For each predictor listed on the y-axis, the observed standardized mean difference is represented by green circles for matched controls and by blue X symbols for all COVID-19 patients in the Mount Sinai Health System (n=4,152). Between plasma recipients and matched controls, all treatment predictors had a standardized mean difference of less than 0.2, represented by the blue shading.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 3 and Supplementary Text 1.
Supplementary Table 2
Oxygen supplementation requirements on days 1, 7 and 14 and end-of-study outcomes for convalescent plasma recipients and matched controls.
Source data
Source Data Fig. 1
Excel table and statistical source data.
Source Data Fig. 2
Excel table and statistical source data.
Source Data Fig. 3
Excel table and statistical source data.
Rights and permissions
About this article

Cite this article
Liu, S.T.H., Lin, HM., Baine, I. et al. Convalescent plasma treatment of severe COVID-19: a propensity score–matched control study. Nat Med 26, 1708–1713 (2020). https://doi.org/10.1038/s41591-020-1088-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-1088-9
- Springer Nature America, Inc.