

Abstract
Bimetallic nanoparticles afford geometric variation and electron redistribution via strong metal-metal interactions that substantially promote the activity and selectivity in catalysis. Quantitatively describing the atomic configuration of the catalytically active sites, however, is experimentally challenged by the averaging ensemble effect that is caused by the interplay between particle size and crystal-phase at elevated temperatures and under reactive gases. Here, we report that the intrinsic activity of the body-centered cubic PdCu nanoparticle, for acetylene hydrogenation, is one order of magnitude greater than that of the face-centered cubic one. This finding is based on precisely identifying the atomic structures of the active sites over the same-sized but crystal-phase-varied single-particles. The densely-populated Pd-Cu bond on the chemically ordered nanoparticle possesses isolated Pd site with a lower coordination number and a high-lying valence d-band center, and thus greatly expedites the dissociation of H2 over Pd atom and efficiently accommodates the activated H atoms on the particle top/subsurfaces.
Similar content being viewed by others
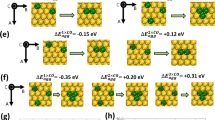

Introduction
Bimetallic catalysts, consisting of a platinum-group metal and a late-transition metal, featured geometrical variation and electronic redistribution via metal–metal interactions and enhanced the activity and/or selectivity, strongly depending on the crystal-phase and the particle size19 or small amount (0.02–2.0 mol.%) of Pd on Cu particles (2–40 nm)5,24. PdCu bulk alloys with a majority of Pd (50–90 mol.%), typically adopting the fcc phase, promoted the selectivity towards ethylene but at the expense of activity46,57. Pd atoms served as the active sites for dissociating H2 while Cu atoms simply diluted Pd ensembles but did not interact electronically with Pd. Here, the B2 particle is characterized by the densely-populated surface Pd–Cu bond that possessed isolated Pd site with a lower coordination number, and thereby promoted the activity pronouncedly at room temperature under a stoichiometric feed gas condition. To some extents, it circumvented the problems encountered by the newly emerged single-atom alloys and the traditional bulk alloys for the selective hydrogenation of multiple carbon–carbon bonds.
In summary, the comprehensive experimental data and theoretical calculations justified that tuning the crystal-phase of bimetallic catalysts at the single-nanoparticle scale changed the atomic structure of the active sites. The densely-populated Pd–Cu bond on the chemically ordered B2 particle, featured by the low coordination numbers and the high-lying metal d-band centers, greatly facilitated H2 dissociation on the Pd atom and efficiently accommodated the activated H atoms on the top/subsurfaces, yielding a much higher activity. This finding fundamentally offers a new route to precisely tailor the geometric and electronic structures of the active sites over bimetallic particles for quantifying the structure−reactivity relationship at atomic accuracy, and practically provides an accessible approach to essentially promote the efficiency of metal catalysts by tuning their intrinsic activity via crystal-phase control at single-nanoparticle level.
Methods
Crystal-phase mediation of PdCu particles
Monodisperse PdCu colloids were prepared by reducing palladium and copper cations with ethylene glycol and using oleylamine as the cap** agent. 66.2 mg Na2PdCl4, 46.9 mg CuCl2·2H2O and 1 ml oleylamine were dissolved into 100 ml ethylene glycol at room temperature under Ar. The mixture was heated to 393 K under stirring and maintained at that temperature for 20 min; further heated to 473 K and kept at that temperature for 2 h. The PdCu colloids were dispersed into cyclohexane (250 ml).
Each PdCu colloid was then precisely coated with a thin silica shell using a water-cyclohexane reverse microemulsion method. PdCu colloids (37.4 mg), dispersed in cyclohexane (250 ml), were mixed with Triton X-100 (polyethylene glycol tert-octylphenyl ether, 80 ml) and ultra-sonically treated for 0.5 h at 293 K. Aqueous ammonia solution (29.4 wt.%, 5 ml) and aqueous hydrazine hydrate solution (80 wt.%, 5 ml) were added to mediate the pH value of the mixture to 13. 9.35 g tetraethyl orthosilicate, mixed with 250 ml cyclohexane, was added and the suspension was stirred for 1 h. The solid product was collected by centrifugation, washed with ethanol, and dried at 323 K under vacuum for 12 h, yielding silica-coated PdCu colloid.
Crystal-phase tuning was done by treating the silica-coated PdCu colloid with reactive gases (H2/O2) at 673–773 K. The ordered body-centered cubic (B2) phase, donated as B2 particle, was prepared by treating the silica-coated PdCu colloid with H2 at 673 K for 2 h. The face-centered cubic (fcc) phase, named as fcc particle, was obtained by calcining the B2 particle at 673 K in air for 4 h, followed by H2 reduction at 773 K for 2 h.
N2 adsorption–desorption isotherms revealed that the specific surface area was 177 m2 g−1 for the B2 particle while 161 m2 g−1 for the fcc particle, mainly contributed by the porous silica shell generated during the high-temperature treatments under the reactive gases (H2/O2).
STEM/ETEM analysis
STEM images were acquired over a JEOL-ARM 300 F microscope at 300 kV. Energy dispersive X-ray spectroscopy elemental map**s over the particles were collected using a JED-2300 T spectrometer. Environmental TEM observations on the particles under H2 and/or H2/C2H2 were done using an aberration-corrected Titan Themis ETEM G3 microscope at 300 kV. The sample was dispersed into ethanol, and the suspension was deposited onto a thermal E-chip that is equipped with a thin silicon nitride membrane. The sample was pretreated with H2 at 673 K for 30 min. The images were acquired at 303 K and under 1 mbar H2 or H2/C2H2 (molar ratio of 1/1). Image simulation was done using the JEMS software package and the Multi-slice module.
XAS spectra
XAFS spectra of Cu and Pd K-edges were measured at the BL14W1 beamline at Shanghai Synchrotron Radiation Facility, China. The sample (100–250 mg) was pressed into a self-supported wafer and mounted into a reaction cell, where it was treated with a 5.0 vol.% H2/N2 mixture (50 ml min−1) at 673 K for 1 h. The spectra were then recorded at 343 K. EXAFS data were processed according to the standard procedure using Athena and Artemis modules of the IFEFFIT software packages.
IR spectra
IR experiments were done with a dedicated ultrahigh vacuum (UHV) apparatus, combing a FTIR spectrometer (Bruker Vertex 80 v) and a multi-chamber UHV system (Prevac). 200 mg sample was pressed into an inert metal mesh and mounted on a sample holder. The sample was treated with H2 at 673 K for 1 h, exposed to CO at 110 K and gradually heated to 460 K at a rate of 3 K min−1. IR data were accumulated by recording 1024 scans with a resolution of 4 cm−1. Peak fittings on the spectra of CO adsorbed on Pd and Cu sites were performed by the Gaussian function.
Theoretical analysis
DFT calculations were done using the Vienna ab initio Simulation Package (VASP) code. To model the B2 and fcc particles, a 4-layer (3 × 2) B2(110) and a 4-layer (1 × 2) fcc (111) slabs were used. The bottom 2 layers were fixed in their bulk position while other metal atoms and adsorbates allowed to relax. The lattice constants of B2 and fcc phases were calculated to be 3.016 and 3.806 Å, respectively. The transition states of H2 dissociation were located using the dimer method and the convergence criteria were set to 0.05 eV Å−1. Additional computational details are reported in the Supplementary Methods.
Catalytic tests
Selective hydrogenation of acetylene was conducted with a continuous-flow fixed-bed quartz tubular reactor (inner diameter, 6 mm) at atmospheric pressure. 50 mg catalyst (40–60 mesh) was pretreated with a 5.0 vol.% H2/N2 mixture (50 ml min−1) at 673 K for 1 h. After being cooled down to room temperature (298 K), the catalyst was exposed to the reaction gas (1.0 vol.% C2H2/0.5–2.0 vol.% H2/He, 50 ml min−1) that was introduced via a mass flow controller. The outlet from the reactor was analyzed online using a gas chromatograph equipped with a thermal conductivity detector and a flame ionization detector. The reaction rate was measured by controlling the conversion of acetylene below 20% via varying the flow rate of the reaction gas or the weight of the catalyst. The activation energy was measured in the temperature range 268–318 K at H2/C2H2 ratio of 1/1–2/1. The reaction orders with respect to hydrogen and acetylene were determined at 298 K by adjusting the concentrations of H2 (0.5–3.0 vol.%) and C2H2 (1.0–3.0 vol.%) in the feed gases.
Data availability
Source data that support the findings of this study are provided in this paper, which can also be available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Gilroy, K. D., Ruditskiy, A., Peng, H. C., Qin, D. & **a, Y. Bimetallic nanocrystals: syntheses, properties, and applications. Chem. Rev. 116, 10414–10472 (2016).
Fan, Z. & Zhang, H. Crystal phase-controlled synthesis, properties and applications of noble metal nanomaterials. Chem. Soc. Rev. 45, 63–82 (2016).
Wong, A., Liu, Q., Griffin, S., Nicholls, A. & Regalbuto, J. R. Synthesis of ultrasmall, homogeneously alloyed, bimetallic nanoparticles on silica supports. Science 358, 1427–1430 (2017).
Li, J. & Sun, S. Intermetallic nanoparticles: synthetic control and their enhanced electrocatalysis. Acc. Chem. Res. 52, 2015–2025 (2019).
Hannagan, R. T., Giannakakis, G., Flytzani-Stephanopoulos, M. & Sykes, E. C. H. Single-atom alloy catalysis. Chem. Rev. 120, 12044–12088 (2020).
Ryoo, R. et al. Rare–earth–platinum alloy nanoparticles in mesoporous zeolite for catalysis. Nature 585, 221–224 (2020).
Chen, Y. et al. Phase engineering of nanomaterials. Nat. Rev. Chem. 4, 243–256 (2020).
Zhou, M., Li, C. & Fang, J. Noble-metal based random alloy and intermetallic nanocrystals: syntheses and applications. Chem. Rev. 121, 736–795 (2021).
Dasgupta, A. et al. Atomic control of active-site ensembles in ordered alloys to enhance hydrogenation selectivity. Nat. Chem. 14, 523–529 (2022).
Wang, C., Chen, D. P., Sang, X., Unocic, R. R. & Skrabalak, S. E. Size-dependent disorder–order transformation in the synthesis of monodisperse intermetallic PdCu nanocatalysts. ACS Nano 10, 6345–6353 (2016).
Ma, S. et al. Electroreduction of carbon dioxide to hydrocarbons using bimetallic Cu–Pd catalysts with different mixing patterns. J. Am. Chem. Soc. 139, 47–50 (2017).
Qiu, Y. et al. BCC-phased PdCu alloy as a highly active electrocatalyst for hydrogen oxidation in alkaline electrolytes. J. Am. Chem. Soc. 140, 16580–16588 (2018).
Tong, W. et al. Crystal-phase-engineered PdCu electrocatalyst for enhanced ammonia synthesis. Angew. Chem. Int. Ed. 59, 2649–2653 (2020).
Li, X. et al. No annealing synthesis of ordered intermetallic PdCu nanocatalysts for boosting formic acid oxidation. Chem. Mater. 34, 1385–1391 (2022).
Ji, Y. et al. Selective CO-to-acetate electroreduction via intermediate adsorption tuning on ordered Cu–Pd sites. Nat. Catal. 5, 251–258 (2022).
Friedrich, M., Villaseca, S. A., Szentmiklósi, L., Teschner, D. & Armbrüester, M. Order-induced selectivity increase of Cu60Pd40 in the semi-hydrogenation of acetylene. Materials 6, 2958–2977 (2013).
Rochefort, A., Abon, M., Delichère, P. & Bertolini, J. C. Alloying effect on the adsorption properties of Pd50Cu50 {111} single crystal surface. Surf. Sci. 294, 43–52 (1993).
Lopez, N. & Nørskov, J. K. Synergetic effects in CO adsorption on Cu-Pd(111) alloys. Surf. Sci. 477, 59–75 (2001).
Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012).
Romas, M., Martínez, A. E. & Busnengo, H. F. H2 dissociation on individual Pd atoms deposited on Cu(111). Phys. Chem. Chem. Phys. 14, 303–310 (2012).
Boucher, M. B. et al. Single atom alloy surface analogs in Pd0.18Cu15 nanoparticles for selective hydrogenation reactions. Phys. Chem. Chem. Phys. 15, 12187–12196 (2013).
Kim, S. K., Lee, J. H., Ahn, I. Y., Kim, W. J. & Moon, S. H. Performance of Cu-promoted Pd catalysts prepared by adding Cu using a surface redox method in acetylene hydrogenation. Appl. Catal. A 401, 12–19 (2011).
Cao, X., Mirjalili, A., Wheeler, J., **e, W. & Jang, B. W. L. Investigation of the preparation methodologies of Pd-Cu single atom alloy catalysts for selective hydrogenation of acetylene. Front. Chem. Sci. Eng. 9, 442–449 (2015).
McCue, A. J. & Anderson, J. A. CO induced surface segregation as a means of improving surface composition and enhancing performance of CuPd bimetallic catalysts. J. Catal. 329, 538–546 (2015).
Marakatti, V. S., Sarma, S. C., Joseph, B., Banerjee, D. & Peter, S. C. Synthetically tuned atomic ordering in PdCu nanoparticles with enhanced catalytic activity toward solvent-free benzylamine oxidation. ACS Appl. Mater. Interfaces 9, 3602–3615 (2017).
Pei, G. X. et al. Performance of Cu-alloyed Pd single-atom catalyst for semi-hydrogenation of acetylene under simulated front-end conditions. ACS Catal. 7, 1491–1500 (2017).
Friedrich, M. & Armbrüster, M. Crystallite size controls the crystal structure of Cu60Pd40 nanoparticles. Chem. Mater. 21, 5886–5891 (2009).
Yamauchi, M. & Tsukuda, T. Production of an ordered (B2) CuPd nanoalloy by low-temperature annealing under hydrogen atmosphere. Dalton Trans. 40, 4842–4845 (2011).
Mukundan, V. et al. Nanoalloying and phase transformations during thermal treatment of physical mixtures of Pd and Cu nanoparticles. Sci. Technol. Adv. Mater. 15, 025002 (1–16) (2014).
Li, G. et al. An ordered bcc CuPd nanoalloy synthesised via the thermal decomposition of Pd nanoparticles covered with a metal-organic framework under hydrogen gas. Chem. Commun. 50, 13750–13753 (2014).
Li, J., Li, L., Guo, S. X., Zhang, J. & Ma, J. PdCu@Pd nanocube with Pt-like activity for hydrogen evolution reaction. ACS Appl. Mater. Interfaces 9, 8151–8160 (2017).
Wu, Z. P. et al. Revealing the role of phase structures of bimetallic nanocatalysts in the oxygen reduction reaction. ACS Catal. 8, 11302–11313 (2018).
Hwang, B. J. et al. Structural models and atomic distribution of bimetallic nanoparticles as investigated by X-ray absorption spectroscopy. J. Am. Chem. Soc. 127, 11140–11145 (2005).
Bozzolo, G., Garcés, J. E., Noebe, R. D., Abel, P. & Mosca, H. O. Atomistic modeling of surface and bulk properties of Cu, Pd and the Cu–Pd system. Prog. Surf. Sci. 73, 79–116 (2003).
Long, R. et al. Isolation of Cu atoms in Pd lattice: forming highly selective sites for photocatalytic conversion of CO2 to CH4. J. Am. Chem. Soc. 139, 4486–4492 (2017).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009).
Armbrüster, M. et al. Pd-Ga intermetallic compounds as highly selective semi-hydrogenation catalysts. J. Am. Chem. Soc. 132, 14745–14747 (2010).
Matselko, O. et al. Revealing electronic influences in the semi-hydrogenation of acetylene. J. Phys. Chem. C. 122, 21891–21896 (2018).
Hoffmann, F. M. & Paul, J. A FT‐IRAS study of the vibrational properties of CO adsorbed on Cu/Ru(001). II. the dispersion of copper. J. Chem. Phys. 87, 1857–1865 (1987).
Kruppe, C. M., Krooswyk, J. D. & Trenary, M. Polarization-dependent infrared spectroscopy of adsorbed carbon monoxide to probe the surface of a Pd/Cu(111) single-atom alloy. J. Phys. Chem. C. 121, 9361–9369 (2017).
Lear, T. et al. The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J. Chem. Phys. 123, 174706 (1–13) (2005).
Rupprechter, G. Sum frequency generation and polarization-modulation infrared reflection absorption spectroscopy of functioning model catalysts from ultrahigh vacuum to ambient pressure. Adv. Catal. 51, 133–263 (2007).
Kohler, M. A., Cant, N. W., Wainwright, M. S. & Trimm, D. L. Infrared spectroscopic studies of carbon monoxide adsorbed on a series of silica-supported copper catalysts in different oxidation states. J. Catal. 117, 188–201 (1989).
Teschner, D. et al. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 320, 86–89 (2008).
Crespo-Quesada, M., Cárdenas-Lizana, F., Dessimoz, A. L. & Kiwi-Minsker, L. Modern trends in catalyst and process design for alkyne hydrogenations. ACS Catal. 2, 1773–1786 (2012).
Zhang, L., Zhou, M., Wang, A. & Zhang, T. Selective hydrogenation over supported metal catalysts: from nanoparticles to single atoms. Chem. Rev. 120, 683–733 (2020).
Yang, B., Burch, R., Hardacre, C., Headdock, G. & Hua, P. Influence of surface structures, subsurface carbon and hydrogen, and surface alloying on the activity and selectivity of acetylene hydrogenation on Pd surfaces: a density functional theory study. J. Catal. 305, 264–276 (2013).
Li, X. T., Chen, L., Wei, G. F., Shang, C. & Liu, Z. P. Sharp increase in catalytic selectivity in acetylene semihydrogenation on Pd achieved by a machine learning simulation-guided experiment. ACS Catal. 10, 9694–9705 (2020).
Shi, X. et al. Copper catalysts in semihydrogenation of acetylene: from single atoms to nanoparticles. ACS Catal. 10, 3495–3504 (2020).
Zhang, T., Walsh, A. G., Yu, J. & Zhang, P. Single-atom alloy catalysts: structural analysis, electronic properties and catalytic activities. Chem. Soc. Rev. 50, 569–588 (2021).
López, N. & Vargas-Fuentes, C. Promoters in the hydrogenation of alkynes in mixtures: insights from density functional theory. Chem. Commun. 48, 1379–1391 (2012).
Fu, Q. & Luo, Y. Catalytic activity of single transition-metal atom doped in Cu(111) surface for heterogeneous hydrogenation. J. Phys. Chem. C. 117, 14618–14624 (2013).
Zhang, R. et al. Insight into the effects of Cu component and the promoter on the selectivity and activity for efficient removal of acetylene from ethylene on Cu-based catalyst. J. Phys. Chem. C. 121, 27936–27949 (2017).
Yang, K. & Yang, B. Surface restructuring of Cu-based single-atom alloy catalysts under reaction conditions: the essential role of adsorbates. Phy. Chem. Chem. Phy. 19, 18010–18017 (2017).
Darby, M. T., Stamatakis, M., Michaelides, A. & Sykes, E. C. H. Lonely atoms with special gifts: breaking linear scaling relationships in heterogeneous catalysis with single-atom alloys. J. Phys. Chem. Lett. 9, 5636–5646 (2018).
Jørgensen, M. & Grönbeck, H. Selective acetylene hydrogenation over single-atom alloy nanoparticles by kinetic Monte Carlo. J. Am. Chem. Soc. 141, 8541–8549 (2019).
Louis, C. & Delannoy, L. Selective hydrogenation of polyunsaturated hydrocarbons and unsaturated aldehydes over bimetallic catalysts. Adv. Catal. 64, 1–88 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21573221, Y.L.; 21533009, W.S.; U1832174, Y.Z.; 91945302, W.L.) and the Deutsche Forschungsgemeinschaft of Germany (392178740, 426888090, Y.W.).
Author information
Authors and Affiliations
Contributions
S.L., Y.L. and W.S. prepared the catalysts and performed the reaction tests. X.Y., Y.W. and C.W. did IR experiments and data analysis. Y.Y., H.Z. and J.Z. conducted XAS measurements and strucutre simulations. C.Z. and W.L. did DFT calculations. S.H. and Y.Z. collected STEM/ETEM images and performed image simulations. Y.L., Y.W., W.L., C.W. and W.S. designed the experiments, discussed the data and wrote the paper. All of the authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, S., Li, Y., Yu, X. et al. Tuning crystal-phase of bimetallic single-nanoparticle for catalytic hydrogenation. Nat Commun 13, 4559 (2022). https://doi.org/10.1038/s41467-022-32274-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32274-4
- Springer Nature Limited