

Abstract
Low thermal conductivity is favorable for preserving the temperature gradient between the two ends of a thermoelectric material, in order to ensure continuous electron current generation. In high-performance thermoelectric materials, there are two main low thermal conductivity mechanisms: the phonon anharmonic in PbTe and SnSe, and phonon scattering resulting from the dynamic disorder in AgCrSe2 and CuCrSe2, which have been successfully revealed by inelastic neutron scattering. Using neutron scattering and ab initio calculations, we report here a mechanism of static local structure distortion combined with phonon-anharmonic-induced ultralow lattice thermal conductivity in α-MgAgSb. Since the transverse acoustic phonons are almost fully scattered by the compound’s intrinsic distorted rocksalt sublattice, the heat is mainly transported by the longitudinal acoustic phonons. The ultralow thermal conductivity in α-MgAgSb is attributed to its atomic dynamics being altered by the structure distortion, which presents a possible microscopic route to enhance the performance of similar thermoelectric materials.
Similar content being viewed by others
Introduction
Transverse acoustic phonons are believed to compete with structure disorder, such as in superionic crystals1,2, glasses3, liquids4, and model crystal-like aperiodic solids5. The main heat carriers in thermoelectric materials are acoustic phonons1,6. Thermoelectric materials, which can be used to directly convert thermal energy and electrical energy, have attracted much attention for meeting current and future energy demands6,7,8,9. The thermoelectric conversion efficiency is governed by the material’s figure of merit7, ZT = [S2σ/(κlat + κele)]T, where S, σ, κlat, κele, and T are the Seebeck coefficient, electronic conductivity, lattice thermal conductivity, electronic thermal conductivity, and absolute temperature, respectively. Low thermal conductivity is one of the most vital properties of high-performance thermoelectric materials1,7,10,11. Besides band engineering, which can enhance a material’s electrical transport properties, many manipulations have been studied with the aim of modulating the κlat, including nano-crystallization12,13,14, crystal defects15,16, structure disorder1,17, rattling guest-filling18,19 among other techniques, which are beneficial for increasing phonon scattering1,7,13,20. On the other hand, materials with intrinsically low κlat, such as PbTe10,21, SnSe22,23, BiSe24, skutterudites18, Bi2Te325, MCrSe2 (M = Ag or Cu)1,2, and MgAgSb15, are of great interest11. Thus, it is both scientifically and technologically significant to study the structure and atomic dynamics of high-performance thermoelectric materials.
The high-performance MgAgSb-based thermoelectric materials have great potential as candidates for near-room temperature (RT) thermoelectric generators15,26. Their ZT values reach ~0.9 at 300 K, and a maximum of 1.4 at 453 K, which fills the materials gap between low-temperature Bi2Te3 alloys and the middle-temperature PbTe systems in the ZT spectrum10,15,25,27. A record high thermoelectric conversion efficiency of 8.5% with a single thermoelectric leg operating at between 293 and 518 K has been achieved28. Recently, an improved ZT of 2.0 and conversion efficiency of 12.6% in Zr- and Pd-doped MgAgSb p-type materials were predicted theoretically29. The κlat of these materials is 0.4 ~0.5 Wm−1K−1, comparable with that of the ultralow κlat in SnSe originated from the strong lattice anharmonicity22,27. This material has three types of structures at different temperatures, the half-Heusler structure γ-MgAgSb at high temperatures >633 K, the Cu2Sb-related structure β-MgAgSb at intermediate temperatures between 633 and 563 K, and the tetragonal structure α-MgAgSb at low temperatures between 563 and 303 K30. A distorted Mg–Sb rocksalt-type sublattice can be formed in α-MgAgSb30. Study of the detailed structure and dynamics of the MgAgSb-based materials is vital to understand the origin of their high-performance thermoelectric properties, with emphasis on their low κlat1,10,12,27,31.
Thus far, in addition to transport property measurements, experimental characterizations of the MgAgSb-based materials have been mainly based on X-ray diffraction and electron microscopy, which have provided microscopic insight into their crystalline structures15,26,30,32. In contrast, numerous theoretical characterizations have focused on their local crystal structure, electronic band structure, chemical bonding, and atomic dynamics, etc.26,27,29,33. Additionally, to date, there have only been theoretical calculations of the phonon modes for the MgAgSb family29,34 without any experimental verification. A detailed atomistic understanding of the ultralow κlat in α-MgAgSb has remained elusive due to the lack of such phonon measurements. Fortunately, new chopper spectrometers at state-of-the-art high-flux neutron sources coupled with advances in high-resolution neutron instruments have enabled high-precision measurements of the dynamic structure factor, S(Q, E), which contains information on the atomic dynamics1,2,10,23.
Here, we use neutron scattering measurements together with systematic ab initio simulations to study the crystalline structure and the atomic dynamics of two α-MgAgSb-based materials, MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 (the sample of Ni p-type substitution for Ag has a higher anomalous electrical resistivity15), the α-phase of which exhibits the highest thermoelectric performance32. We find that their ultralow κlat is induced both by static local structure distortion suppression of the transverse acoustic phonons and the phonon anharmonicity.
Results
Crystallographic structure properties
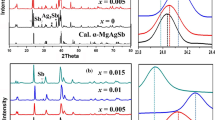
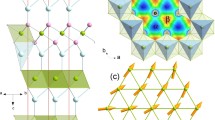
The evolution of the crystallographic structures of MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 as a function of temperature was investigated by neutron diffraction. Both compounds maintain the α-MgAgSb structure (Fig. 1a) over a wide temperature range from 20 to 500 K (Fig. 1b, c and Supplementary Fig. 1a, b). The structure features a 24-atom trigonal primitive unit cell with a large distorted rocksalt sublattice, where the Ag atoms fill half of the Mg–Sb distorted cubes. This structure favors the low thermal conductivity paradigm of crystals with complex unit cells17. The distorted structure has a significant phonon scattering effect and the complex primitive unit cell has a large ratio of optical phonon branches (69/72) that significantly reduces κlat17, since acoustic phonons are the main contributor to κlat1,6,20.

a Crystalline structure of α-MgAgSb with a complex distorted Mg–Sb rocksalt sublattice, where half of the Mg–Sb distorted cubes are filled with Ag atoms. b Rietveld refinement of MgAg0.965Ni0.005Sb0.99 neutron diffraction data measured at 300 K. c Temperature-dependent MgAg0.965Ni0.005Sb0.99 neutron diffraction data. These data reveal that MgAg0.965Ni0.005Sb0.99 maintains the same α-MgAgSb tetragonal structure with a space group of I−4c2 (no.120) over a temperature range from 20 to 500 K. The 450 and 500 K data show the appearance of a small amount of Sb precipitate; a.u., arbitrary units.
The temperature-dependent neutron diffraction data (Fig. 1c and Supplementary Fig. 1b) were analyzed by the method of Rietveld refinement, and the lattice thermal dilatation shows anisotropic features as revealed by the increasing value of c/a with increasing temperature (Supplementary Fig. 1f). The lattice thermal expansion has an inflection point at ~200 K (Supplementary Fig. 1c, d), which is consistent with the previously measured thermal conductivity and ZT data27. This indicates that an additional phonon-scattering mechanism related to the structure evolution may appear at high temperatures.
Local structure distortion
Here, we study the local structure of MgAg0.965Ni0.005Sb0.99 using neutron total scattering. By the pair distribution function (PDF) analysis and fitting of G(r), which is a Fourier transform result of the static structure factor, S(Q) (see Methods section)1,20,35, we directly reveal the distortion of the Mg–Sb rocksalt sublattice in real space by the shoulder peak at ~3.4 Å and the small peak at ~5.8 Å (Fig. 2a). The refined Mg–Sb bond distances are in the range from 2.86(2) Å to a large value of 3.90(2) Å, which is larger than the radius summation of Mg and Sb atoms (Mg: 1.60 Å, Sb: 1.44 Å; Fig. 2b). It reveals a strong distortion and weak bonding nature of Mg–Sb bonds, which indicates that novel atomic dynamics may exist23.

a Real space G(r) fitting demonstrates that the first sharp peak at r ~3.0 Å mainly results from bonds between distinct-atoms, while the shoulder peak at r ~4.5 Å mainly results from bonds between atoms of the same elements. The distortion of the Mg–Sb rocksalt results in the split of Mg–Sb nearest-neighbor bonds. Here, this PDF data directly indicates this distortion by the peaks at r ~3.3 and ~5.8 Å (yellow-tinted regions), as a result of the distinguishable contribution of Mg–Sb bonds to these peaks. The data are measured at 300 K. b Distorted structure of the Ag atom-filled Mg–Sb rocksalt sublattice. The refined nearest Mg–Ag bond distances d1–d6 are between 2.86(2) and 3.90(2) Å.
Transverse acoustic phonon suppression from local structure distortion
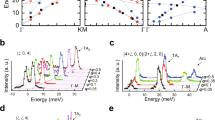
Consequently, we investigated the atomic dynamic properties of these materials by inelastic neutron scattering (INS). Figure 3a shows a representative data set of the Bose-factor-calibrated dynamic structure factor, \(B\left( {{\mathbf{Q}},E} \right) = \left[ {1 - e^{ - \frac{E}{{k_{\mathrm{B}}T}}}} \right]S({\mathbf{Q}},E)\), for the MgAg0.965Ni0.005Sb0.99 compound, obtained with incident neutron energy of Ei = 15.16 meV at 300 K (see Methods section). The S(Q) data for the same sample measured at 300 K were also plotted at the bottom of Fig. 3a for comparison. The intensity of the sharpest peak at Q ~2.76 Å−1 for S(Q) is about four times as strong as the second sharpest one at Q ~2.17 Å−1. This can be ascribed to the particular distorted crystal structure, which has many equivalent Brillouin zone centers in different reciprocal space directions folded at this sharpest peak Q position. We consider these centers to be a quasi-Brillouin-zone (QBZ) center. Thus, a novel double-mushroom scattering pattern arising from the QBZ center is observed in Fig. 3a. To examine the acoustic nature of the low-energy vibration modes, we using the resonant ultrasound spectrometer measured sound velocities, with VT = 1102 m/s and VL = 3708 m/s27, calculated the dispersions that are the magenta and green solid lines arising from the QBZ center in Fig. 3a for the transverse and longitudinal acoustic modes, respectively. The E-cut data at 2.0, 2.5, and 3.0 meV are shown in Fig. 3d. The green and magenta arrows indicate the longitudinal and transverse phonon peak positions, respectively, which were calculated using the sound velocities. The ab initio simulation result, using the VASP and OCLIMAX programs (see Methods section), is shown in Fig. 3b. By linking together the INS experimental results, the simulation results (Fig. 3b and Supplementary Fig. 2), and the calculations by the sound velocities, and applying them to the observed double-mushroom scattering pattern (Fig. 3a), it was determined that the lower branches at E ~4.5 meV are mainly the longitudinal acoustic phonon modes, whereas the upper branches at E ~7.0 meV are mainly the low-energy optical phonon modes (Figs. 3a, 4b). Most importantly, these results illustrate that the transverse acoustic phonons are fundamentally suppressed in this material.

a Bose-factor-calibrated dynamic structure factor, B(Q, E), measured with INS (top) and static structure factor, S(Q), measured with neutron total scattering (bottom) at 300 K in MgAg0.965Ni0.005Sb0.99. The magenta and green lines are calculated dispersions based on transverse and longitudinal sound velocities, respectively, which were measured by the resonant ultrasound spectrometer method. b The corresponding neutron-weighted ab initio calculated B(Q, E) pattern using the OCLIMAX program for α-MgAgSb. c The corresponding neutron-weighted ab initio calculated B(Q, E) pattern based on an adjusted symmetric Ag atom-filled Mg–Sb rocksalt structure. d E-cut data at 2.0, 2.5, and 3.0 meV. The magenta and green arrows indicate the transverse and the longitudinal phonon peak positions, respectively, which are calculated by the sound velocities. Error bars are propagated from counting statistics on measured spectra; a.u., arbitrary units. e A schematic shows transverse acoustic phonon suppression by the static local structure distortion. f The adjusted symmetric Ag atom-filled Mg–Sb rocksalt structure used in c calculation. By comparing b with c, the transverse acoustic phonons are observed to survive in the symmetric structure while they disappear in the distorted structure e. These results demonstrate that the transverse acoustic phonons are mostly suppressed by the distorted structure in this material. For the purpose of comparison, the color bars in a, b, and c are plotted in relative intensities with arbitrary units.

a Neutron-weighted GPDOS measured by INS in MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 at 5 K and the ab initio calculations for the α-MgAgSb tetragonal phase at 0 K. b Neutron-weighted low-energy BPDOS measured by high-resolution INS in MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 at selected temperatures. Here, the low-energy peak at ~4.5 meV is mainly due to acoustic phonon modes and the peak at ~7.0 meV is mainly due to low-frequency optical phonon modes. c Temperature dependence of the peak positions of acoustic phonons and low-frequency optical phonons, which are fitted from INS data shown in b, showing gradual softening. The phonon modes of the Ni-doped sample are much softer than those of the parent sample. The lines are the linear fitting results. The values of the phonon softening ratio (the slope of the fitting lines) are shown in Supplementary Table 3. Error bars in a and b are propagated from counting statistics on measured spectra and error bars in c are result from the statistical uncertainties in fitting the phonon peaks; a.u., arbitrary units.
To study the origin of the suppression of the transverse acoustic phonons, we adjusted the distorted Mg–Sb sublattice to a highly symmetric Mg–Sb rocksalt structure by making all of the Mg–Sb bonds equivalent (Fig. 3f). We then calculated the phonon spectrum of this non-distorted structure (Supplementary Fig. 3) and the corresponding scattering pattern (Fig. 3c), using the same method and instrument parameters as in Fig. 3b. Here, the longitudinal and transverse acoustic branches marked as L and T in Fig. 3c, respectively, arising from the QBZ center, i.e., Q ~2.76 Å−1, can be clearly seen. Magnifications of areas of B(Q, E) at the Q from 2.2 to 3.3 Å−1 and E from 0 to 10 meV region in Fig. 3a–c are shown in Supplementary Fig. 4, making their differences clearly visible. By comparing these high-symmetry structure results with those of the distorted structure, the suppression of the transverse acoustic phonons by the static local structure distortion in α-MgAgSb is clear (Fig. 3e). We stress that this suppression results in the ultralow κlat of α-MgAgSb, which is different from the case of superionic conductors AgCrSe21 and CuCrSe22, in which the dynamic disorder with crystal structure transition suppresses the transverse acoustic phonons.
Phonon softening
Figure 4 shows the atomic dynamic properties in the MgAgSb-based thermoelectric materials, which are measured by INS and calculated by ab initio calculations (see Methods section). Here, we demonstrate that the values of the generalized phonon density of states (GPDOS) of both MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 measured at 5 K by INS are in good overall agreement with the simulation results of α-MgAgSb, not only in terms of the total shape but also the energy of the main features, including the three main optical phonon peaks at ~8, 14, and 24 meV and the acoustic phonon shoulder peak at ~4.5 meV (Fig. 4a).
Since the ZT value of both MgAg0.97Sb0.99 and MgAg0.965Ni0.005Sb0.99 increases with increasing temperature before reaching a maximum at ~450 K with a plateau from 450 to 550 K15,27, we performed temperature-dependent high-resolution INS measurements to further study the properties of the low-frequency phonons, especially the acoustic phonons. Figure 4b shows the Bose-factor-calibrated phonon density of states (BPDOS) as a function of temperature (see Methods section). Here, the energy gap between the low-energy optical phonons and the acoustic phonons at ~5 meV is mapped much more clearly, benefitting from the higher resolution. More interestingly, a temperature-induced phonon softening is shown by these data. The peak positions of the acoustic phonon modes and the low-energy optical phonon modes fitted by the Gaussian function are plotted in Fig. 4c. The corresponding ab initio calculation results using the temperature-dependent lattice parameters are shown in Supplementary Fig. 5. As the temperature-induced phonon softening mainly arises from the lattice expansion and anharmonicity2, the anharmonic nature of α-MgAgSb can be verified by comparing the INS and simulation results. We find that the softening ratio of the INS-measured BPDOS as a function of temperature is about two times as strong as that of the corresponding simulation (Fig. 4c, Supplementary Fig. 5, Supplementary Table 3), which reveals its anharmonic nature.
Low thermal conductivity mechanism
To further verify the low thermal conductivity mechanism, we computed the intrinsic anharmonic effects of α-MgAgSb and the high-symmetry structure MgAgSb (the γ-MgAgSb shown in Fig. 3f) from first principles using ShengBTE36 and Phonopy37. Figure 5a shows the temperature-dependent κlat of α-MgAgSb demonstrating the overall agreement between the calculation results of MgAgSb and the experimental results of MgAg0.97Sb0.9915,38. The calculated RT κlat of α-MgAgSb is 0.54 Wm−1 K−1, which is comparable with the experimental value of MgAg0.97Sb0.99 (~0.6 Wm−1 K−1)15. As clearly indicated in Fig. 5b, c, the phonon group velocities (v) of α-MgAgSb are lower than those of γ-MgAgSb, while its phonon lifetimes (τ) are larger than those of γ-MgAgSb, especially for the acoustical phonons <10 meV in γ-MgAgSb. The three-phonon process is easier to occur in γ-MgAgSb compared to α-MgAgSb demonstrated by the calculated three-phonon scattering phase space shown in Supplementary Fig. 6, and accordingly enhance the phonon anharmonicity of γ-MgAgSb. The total Grüneisen parameter (γtotal) obtained as a weighted sum of the mode contributions at 300 K are 1.51 and 3.03 for α-MgAgSb and γ-MgAgSb, respectively (Fig. 5d). Generally, large γtotal corresponds to large phonon anharmonicity and low κlat, for instance the γtotal are 1.45 for PbTe38 (Cal.) and ref. 15 (Exp.) are also plotted for comparison. According to κtot = κlat + κele = κlat + LσT, the lattice thermal conductivity (κlat) can be obtained by subtracting κele from the κtot. The shadow regions are the experimental thermal conductivity of MgAg0.97Sb0.99 with the Lorenz number7,15 being from 1.6 to 2.6 × 10−8 WΩK−2. Here, we only deal with the pure α-MgAgSb crystal and consider the phonon–phonon coupling to stimulate the phonon transport properties. Our calculated values are in excellent agreement with the previous reports and our measured values15,38. b Calculated phonon group velocities v, c phonon relaxation time τ, and d Grüneisen parameters γgru for α- and γ-MgAgSb at 300 K. The inset in d shows total γtotal obtained as a weighted sum of the mode contributions, as a function of temperature for α- and γ-MgAgSb. The γgru of high values accumulate in the vicinity of 5 meV corresponding to the transverse acoustic phonon modes for γ-MgAgSb.
