
Abstract
Hepatoma-derived growth factor (HDGF) expression is associated with poor prognosis in non-small cell lung cancer (NSCLC); however, whether HDGF affects gefitinib resistance in NSCLC remains unknown. This study aimed to explore the role of HDGF in gefitinib resistance in NSCLC and to discover the underlying mechanisms. Stable HDGF knockout or overexpression cell lines were generated to perform experiments in vitro and in vivo. HDGF concentrations were determined using an ELISA kit. HDGF overexpression exacerbated the malignant phenotype of NSCLC cells, while HDGF knockdown exerted the opposite effects. Furthermore, PC-9 cells, which were initially gefitinib-sensitive, became resistant to gefitinib treatment after HDGF overexpression, whereas HDGF knockdown enhanced gefitinib sensitivity in H1975 cells, which were initially gefitinib-resistant. Higher levels of HDGF in plasma or tumor tissue also indicated gefitinib resistance. The effects of HDGF on promoting the gefitinib resistance were largely attenuated by MK2206 (Akt inhibitor) or U0126 (ERK inhibitor). Mechanistically, gefitinib treatment provoked HDGF expression and activated the Akt and ERK pathways, which were independent of EGFR phosphorylation. In summary, HDGF contributes to gefitinib resistance by activating the Akt and ERK signaling pathways. The higher HDGF levels may predict poor efficacy for TKI treatment, thus it has the potential to serve as a new target for overcoming tyrosine kinase inhibitor resistance in combating NSCLC.

Similar content being viewed by others


Introduction
Lung cancer is the most common cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) accounts for approximately 80% of all lung carcinoma cases. Patients with epidermal growth factor receptor (EGFR) mutations respond well to tyrosine kinase inhibitors (TKIs), but all responding patients eventually develop acquired resistance to TKIs [1]. The underlying mechanisms of TKI resistance include secondary resistance mutations of targeted genes, activation of bypass signaling pathways, dysregulation of downstream effectors, and phenotypic transformation [2]. T790M mutation in exon 20 of EGFR accounts for approximately 50% of resistant cases and is the predominant resistance mechanism of TKIs. However, approximately 19% of resistance mechanisms remain unknown [3].
Hepatoma-derived growth factor (HDGF) is a heparin-binding growth factor that was first identified from the conditioned medium of the Huh-7 hepatoma cell line [4], indicating that it can be secreted. HDGF has been reported to be involved in cancer cell growth, angiogenesis, anti-apoptosis, and tumor metastasis [4, 5]. In a range of tumor types, including NSCLC, ovarian cancer, oral cancer, gastric cancer and melanoma, HDGF is overexpressed, and its higher expression is strongly correlated with poor prognosis [6,7,8,9,10,11]. HDGF enhances vascular endothelial growth factor (VEGF)-dependent angiogenesis in NSCLC [12], and high serum HDGF levels predict bone metastasis and unfavorable prognosis in NSCLC [13]. In surgically resected stage I NSCLC, HDGF is regarded as a biomarker for molecular staging and prognosis and provides prediction for postoperative adjuvant chemotherapy [14]. Inhibition of HDGF suppressed tumor cell growth both in vitro and in vivo [15, 16]. Antibodies targeting HDGF could prevent NSCLC relapse after chemotherapy by suppressing cancer stem cells [17, 18]. Some microRNAs (miRNAs) inhibit NSCLC proliferation and invasion by targeting HDGF [19,20,Full size image
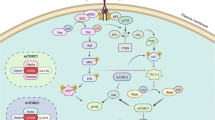
We then asked whether HDGF and EGFR have complementary roles in responding to gefitinib in NSCLC cells with HDGF loss and gain. As shown in Fig. 6G, I, HDGF knockdown caused a decrease in HDGF expression, and p-EGFR upregulation was reversed by gefitinib treatment in H1975 cells. In H292 and PC-9 cells, HDGF overexpression resulted in HDGF elevation and p-EGFR suppression, and gefitinib treatment further inhibited p-EGFR but without an obvious influence on HDGF expression. As shown in Fig. 6H, J, HDGF was increased by gefitinib administration in PC-9 xenograft tumors with HDGF overexpression (P < 0.05), and other changes in HDGF and p-EGFR in tumor tissues were consistent with the cell experiments. Therefore, the above data indicate crosstalk between HDGF and EGFR in NSCLC, and HDGF may serve as a bypass signaling molecule of EGFR to activate the downstream molecules Akt and ERK. Furthermore, HDGF and EGFR have complementary roles in maintaining the survival of tumor cells exposed to gefitinib. The function of HDGF in gefitinib resistance is illustrated by a schematic diagram in Fig. 7.

Schematic diagram of the contribution of HDGF to gefitinib resistance by activating EGFR downstream molecules as a bypass in NSCLC.
