
Abstract
The global burden of colorectal cancer (CRC) has rapidly increased in recent years. Dysregulated cholesterol homeostasis facilitated by extracellular matrix (ECM) remodeling transforms the tumor microenvironment. Collagen I, a major with ECM component is highly expressed in colorectal tumors with infiltrative growth. Although oxysterol binding protein (OSBP)-related proteins accommodate tumorigenesis, OSBPL2, which is usually involved in deafness, is not associated with CRC progression. Therefore, we aimed to investigate the pathological function of OSBPL2 and identify the molecular link between ECM-Collagen I and OSBPL2 in CRC to facilitate the development of new treatments for CRC. OSBPL2 predicted a favorable prognosis in stage IV CRC and substantially repressed Collagen I-induced focal adhesion, migration, and invasion. The reduction of OSBPL2 activated ERK signaling through the VCAN/AREG/EREG axis during CRC growth, while relying on PARP1 via ZEB1 in CRC metastasis. OSBPL2 defect supported colorectal tumor growth and metastasis, which were suppressed by the ERK and PARP1 inhibitors SCH772984 and AG14361, respectively. Overall, our findings revealed that the Collagen I-induced loss of OSBPL2 aggravates CRC progression through VCAN-mediated ERK signaling and the PARP1/ZEB1 axis. This demonstrates that SCH772984 and AG14361 are reciprocally connective therapies for OSBPL2Low CRC, which could contribute to further development of targeted CRC treatment.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is a prevalent malignancy worldwide, with a high incidence and mortality [1]. Population aging has increased the CRC burden in China and the United States of America. This warrants further development of strategies for cancer prevention and treatment [Full size image
OSBPL2 modifies ERK signaling through the VCAN/AREG/EREG axis in Collagen I-induced cell growth but not in EMT
Transcriptome sequencing was performed to investigate the molecular mechanisms underlying OSBPL2 expression in CRC. RNA-seq analysis showed 564 and 668 genes whose expression was upregulated and downregulated, respectively, in OSBPL2 KD HCT116 cells compared to shcontrol cells (Fig. 5A). Proteoglycans in cancer and cell adhesion molecules were the two most enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Fig. 5B). The two most differentially expressed genes were amphiregulin (AREG) and Versican (VCAN) (Table S1). AREG, a member of the epidermal growth factor (EGF) family, regulates mitogen-activated protein kinase (MAPK) signaling. The expression of epiregulin (EREG), another EGF, was upregulated by OSBPL2 knockdown.

A The volcano plots of genes in shcontrol and OSBPL2 KD HCT116 cells analyzed by RNA-seq. B The KEGG enrichment pathways of differential genes followed by knockdown of OSBPL2. C The hypothesis of regulation of VCAN/AREG/EREG by OSBPL2 deficiency. D–G The mRNA expression of VCAN (D) and COL1A1 (F) in CRC tumor tissues and normal tissues (from TCGA database). Tumor samples in COAD were divided into high and low expression groups based on the median VCAN or COL1A1 expression level, and the survival of the two groups was analyzed using the Kaplan–Meier method in all stages (E) and stage II (G). H The correlation between COL1A1 and VCAN in CRC. I The correlation between VCAN and AREG in Stage I COAD (Colorectal adenocarcinoma). J The correlation between VCAN and EREG in Stage I COAD. K The correlation between AREG and EREG in CRC. L The expressions of VCAN/AREG/EREG were examined by real-time PCR in shcontrol and OSBPL2 KD HCT116 cells, ACTB was used as a housekee** gene. M The protein levels of total ERK and Phospho-ERK1/2 (Thr202/Tyr204) in shcontrol and OSBPL2 KD HCT116 cells were measured by western blot. N After knockdown Versican by three VCAN siRNAs, the expressions of AREG were examined by real-time PCR, ACTB was used as a housekee** gene. O The protein levels of total ERK and Phospho-ERK1/2 (Thr202/Tyr204) were measured by western blot, followed by knockdown Versican by three VCAN siRNAs. P, Q The shcontrol and OSBPL2 KD HCT116 were treated with SCH772984 (1.25 nM), respectively, then be detected by colony formation assay.
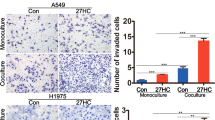
VCAN is a cell adhesion molecule in the ECM that coordinates with EGF to modify MAPK signaling in OSBPL2 KD CRC cells (Fig. 5C). In the TCGA database, the expression of VCAN and COL1A1 was elevated in CRC. In addition, higher COL1A1 levels were associated with poor prognosis in stage II CRC (Fig. 5D–G). Although VCAN was not a prognostic marker in CRC, it positively correlated with COL1A1 (Fig. 5H). Additionally, VCAN correlated positively with AREG and EREG in stage I, and AREG correlated positively with EREG in all stages (Fig. 5I–K). Thus, the expression of AREG, EREG, and VCAN and the activation of MAPK signaling were examined. AREG, EREG, and VCAN levels were enhanced by OSBPL2 knockdown (Fig. 5L). Additionally, OSBPL2 deficiency stimulated ERK phosphorylation and VCAN accumulation following incubation with Collagen I (Fig. 5M). VACN knockdown by effective siRNA-2, 3 eliminated the phosphorylation of ERK and increased AREG levels in OSBPL2 KD CRC cells (Figs. 5N–O and S5A). Furthermore, the ERK inhibitor SCH772984 impaired Collagen I-induced colony formation, but not invasion. This was followed by the loss of OSBPL2 (Figs. 5P, Q and S5B, C). These findings demonstrate that a decrease in OSBPL2 provokes VCAN/AREG/AREG-mediated ERK signaling during CRC cell growth.
OSBPL2 protects against colorectal tumor growth by blocking ERK phosphorylation
We used a xenograft tumor model to assess the function of OSBPL2 in colorectal tumor growth in vivo. As shown in Fig. 6A–C, OSBPL2 deficiency significantly expedited the growth of tumors derived from CRC cells, except for SW620 cells. The average weights and volumes of xenograft tumors in mice injected with shcontrol HCT116, HT29, and LoVo cells reached 0.19 g, 0.18 g, and 0.16 g and 149.53 mm3, 187.18 mm3, and 199.83 mm3, respectively. The average weight and tumor volume of mice injected with OSBPL2 KD CRC cells increased by 169.41%, 100.48%, and 129.24% and 294.50%, 151.24%, and 202.70%, respectively. In contrast, OSBPL2 overexpression reduced the weights and volumes of HCT116 and HT29 cell-derived xenograft tumors by 43.18%, 20.62% and 53.58%, 37.69%, respectively (Fig. 6D–I). Consistent with these observations, Ki67 expression considerably increased after the downregulation of OSBPL2, whereas the upregulation of OSBPL2 markedly decreased Ki67 expression (Fig. 6J–L). The expression levels of VCAN, AREG, and EREG were evaluated in tumors derived from OSBPL2 KD CRC cells (Fig. 6M). OSBPL2 was negatively correlated with VCAN, AREG, and EREG, whereas there was a positive correlation between VCAN, AREG, and EREG in colorectal tumors (Fig. 6N–S). Similarly, ERK phosphorylation was elevated in xenograft tumors derived from OSBPL2 KD CRC cells (Fig. 6T). These results demonstrate that OSBPL2 defect facilitates colorectal growth by activating ERK signaling.

Images (A) and weights (B) of the tumors harvested from nude mice and tumor growth curve (C) of shcontrol and OSBPL2 KD HCT116 (n = 11), HT29 (n = 11), LoVo (n = 11) and SW620 (n = 10) cells. Images (D, G) and weights (E, H) of the tumors harvested from nude mice and tumor growth curve (F, I) of vector and overexpression of OSBPL2-flag in OSBPL2 KD HCT116 (n = 12) and HT29 (n = 10) cells. J–L IHC analysis of Ki67 expression in (A), (D) and (G). Scale bars, 20μm. M. The expressions of OSBPL2/VCAN/AREG/EREG were examined by real-time PCR in xenograft tumors derived by shcontrol and OSBPL2 KD HCT116 cells. N–S The correlation among OSBPL2, VCAN, AREG, EREG in (M) were measured by linear regression and Pearson analysis. T The protein levels of total ERK, Phospho-Erk1/2 (Thr202/Tyr204) and OSBPL2 in xenograft tumors were detected by western blot, β-tubulin was utilized as the loading control. Mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. ns no significance.
The function of OSBPL2 on metastasis is mediated by PARP1 through ZEB1
A co-immunoprecipitation (co-IP) assay and mass spectrometry were used to identify the proteins interacting with OSBPL2. An interacting protein band appeared between 100 and 150 kDa (Fig. 7A), and PARP1 was identified based on the highest score (Fig. 7B). The interaction between OSBPL2 and PARP1 was verified using a co-IP assay (Figs. 7C and S6A). PARP1 plays a multifaceted role in DNA repair, chromatin remodeling, and PARylation (poly [ADP] ribosylation) of various EMT-related proteins such as E-cadherin, Snail1, ZEB1, Twist1, p65, and SMADs [41,42,43]. We constructed a PARP1 expression vector and siRNA and applied the PARP1 specific inhibitor AG14361 to validate the relationship between OSBPL2 and PARP1. The IC50 concentration of AG14361 was 13.33 μM, which suppressed cell proliferation without inducing cell death (Fig. S6B).

A The co-IP experiment of determining the proteins interacted with OSBPL2 (depicted in red arrows). B Identifying the proteins in (A) by mass spectrum. C The co-IP assay showing the interaction between OSBPL2 and PARP1. D–F Transwell assays were performed to assess the migration and invasion abilities of shcontrol and OSBPL2 KD HCT116 cells with treatment of DMSO and AG14361. G–I Transwell assays were carried out to evaluate the migration and invasion ability of vector and OSBPL2 OE HCT116 cells with treatment of DMSO and AG14361. J–L The shcontrol and OSBPL2 KD HCT116 cells were transient transfected with siNC, siPARP1-1 and siPARP1-2, then be analyzed by transwell assay. NC negative control. M The shcontrol and OSBPL2 KD HCT116 cells were transient transfected two PARP1 siRNAs, the expressions of PARP1, OSBPL2 were detected by western blot, sicontrol was used as control. N The shcontrol and OSBPL2 KD HCT116 were treated with AG14361 at concentrations of 5 μM for 6 h or 24 h, PARP1, OSBPL2, ZEB1, E-cadherin were detected by western blot, DMSO was used as control. β-actin was served as loading control. O The shcontrol and OSBPL2 KD HCT116 were treated with AG14361 at concentrations of 1 μM, respectively. Immunofluorescence was carried out to detect invadopodia with stimulation of Collagen I for 24 h. Cortactin were labelled in green, F-actin was labelled in red, and the cell nucleus were stained by DAPI (in blue). Scale bars, 10 μm.
AG14361 blocked the capabilities of migration and invasion induced by OSBPL2 in HCT116 cells (Fig. 7D–I). CRC cells were more sensitive to the PARP1 inhibitor, whereas the level of OSBPL2 was low. Hence, OSBPL2 competes with this inhibitor by trap** PARP1 during CRC metastasis. Knockdown of PARP1 by the two siRNAs markedly inhibited the metastatic function of OSBPL2 (Fig. 7J–M). However, the PARP1 overexpression did not impede the metastatic function of OSBPL2 (Fig. S6C–F). Otherwise, PARP1 had no impact on CRC cell adhesion to Collagen I or on the activation of ERK signaling (Fig. S6G–I).
Moreover, AG14361 blocked the upregulation of ZEB1 earlier and the downregulation of E-cadherin expression late (Fig. 7N). It substantially blocked invadopodia formation induced by OSBPL2 knockdown, which may be related to the phosphorylation of cortactin (Figs. 7O and S6J). These data indicate that PARP1 mediates OSBPL2 function in EMT through ZEB1.
OSBPL2 protects against colorectal tumor metastasis following tail vein and intrasplenic injection
To evaluate the function of OSBPL2 in CRC metastasis in vivo, we used tail vein and intrasplenic injection models. The tail vein injection model showed that the lack of OSBPL2 resulted in more metastatic nodules in the lungs, small intestines, and kidneys (Figs. 8A–D and S7A–F). Although there were no differences in the number of metastatic nodules in the lymph nodes, they were larger in the OSBPL2 KD group (Figs. 8C and S7B). Likewise, colorectal length was slightly shorter in the OSBPL2 KD group (Fig. S7F). The histological morphologies of the metastatic nodules are shown in Fig. 8E–G. Overexpression of OSBPL2 restrained tumor metastasis to the lymph nodes and lungs (Figs. 8H–K and S7G–L).

A–D Mice were dissected after being injected with shcontrol and OSBPL2 KD HT29 (n = 11) cells via the tail vein. Representative anatomical images (A), statistics of tumor metastatic sites (B), differences in lymph node metastasis (C), and differences in small intestine metastasis (D). Representative images and H&E-stained scans of the lungs (E), small intestine (F), and kidneys (G) in OSBPL2 KD HT29 group. H–J Mice were dissected after being injected with vector and OSBPL2-flag OE HT29 (n = 12) cells via the tail vein. The weight of the mice was measured, and their condition was observed every 3-4 days. Representative anatomical images (H), statistics of tumor metastatic sites (I), and differences in lymph node metastasis (J). K Representative images and H&E-stained scans of the lungs in vector HT29 group. The metastatic nodules were marked in black arrows. L After establishing a liver metastasis model of intrasplenic injection with shcontrol and OSBPL2 KD HT29 (n = 14) cells, the mice were dissected. The images of livers were shown. Representative H&E-stained images were scanned for the liver tissues indicated by the black arrows. M Representative H&E-stained images of the small intestine (left) and colorectum (right) of mice injected with OSBPL2 KD HT29 cells. N After establishing a liver metastasis model of intrasplenic injection with vector and OSBPL2 OE HCT116 (n = 9) cells, the mice were dissected. The weight of the mice was measured, and their condition was observed every 3–4 days. The images of livers were shown. Representative H&E-stained images were scanned for the liver tissues indicated by the black arrows.
The intrasplenic injection model showed that OSBPL2 depletion increased the liver metastasis burden (Fig. 8L, S8A–S8C). As shown in Figs. 8M, and S8D, E, mice injected with OSBPL2 KD HT29 cells had more metastatic nodules in the small intestines and larger metastatic nodules in the colorectum. In contrast, OSBPL2 overexpression eliminated liver metastasis tumors (Figs. 8N and S8F–I). These results indicate that OSBPL2 protects against CRC metastasis.
SCH772984 and AG14361 relieve colorectal tumor progression induced by OSBPL2 defect
As ERK signaling and PARP1 play important roles OSBPL2 function, SCH772984 and AG14361 were used to treat CRC characterized by OSBPL2 deficiency. The effect of SCH772984 on CRC growth was evaluated using a xenograft tumor model (Fig. 9A). The ERK inhibitor SCH772984 significantly alleviated colorectal tumor growth induced by OSBPL2 knockdown, with reduced average tumor weights and volumes by 41.19% and 39.24%, respectively (Fig. 9B–D). SCH772984 treatment markedly suppressed the activation of ERK signaling in tumors derived from OSBPL2 KD CRC cells (Fig. 9E). Furthermore, SCH772984 substantially improved the poor survival induced by OSBPL2 knockdown (Fig. 9F). The effect of AG14361, a PARP1 inhibitor, on CRC metastasis was examined using a liver metastasis model (Fig. 9G). AG14361 markedly reduced the number of liver metastatic nodules induced by the loss of OSBPL2 (Fig. 9H). These data suggest that SCH772984 and AG14361 suppressed CRC progression in vivo.

A The flow chart of xenograft tumor model derived from shcontrol and OSBPL2 KD HCT116 cells with treatment of SCH772984. i.p: intraperitoneal injection. Images (B) and weights (C) of the tumors harvested from nude mice and tumor growth curve (D) of shcontrol and OSBPL2 KD HCT116 cells with treatment of SCH772984 respectively, DMSO as control. E The protein levels of total ERK, Phospho-Erk1/2 (Thr202/Tyr204) and OSBPL2 in xenograft tumors(B) were detected by western blot, β-tubulin was utilized as the loading control. F Kaplan–Meier analysis of mouse survival and statistical analyses in A are depicted. G The flow chart of tumor metastasis model derived from shcontrol and OSBPL2 KD HCT116 cells with treatment of AG14361. H The images of livers in (F) were shown. The areas of liver tumors were marked in black dotted line.

