
Abstract
Histone deacetylase 3 (HDAC3) plays a crucial role in chromatin remodeling, which, in turn, regulates gene transcription. Hence, HDAC3 has been implicated in various diseases, including ischemic injury, fibrosis, neurodegeneration, infections, and inflammatory conditions. In addition, HDAC3 plays vital roles under physiological conditions by regulating circadian rhythms, metabolism, and development. In this review, we summarize the current knowledge of the physiological functions of HDAC3 and its role in organ injury. We also discuss the therapeutic value of HDAC3 in various diseases.
Similar content being viewed by others

Introduction
Solid-organ injury is among the leading causes of death globally and significantly impacts the quality of life. Solid-organ injury can be acute injury occurring in the perioperative period or chronic injury caused by long-term stimulation and toxic insult. Acute injury includes myocardial, cerebral, renal, and hepatic ischemia-reperfusion injury (IRI) [1, 2]. Acute organ injury is characterized by potent proinflammatory responses involving leukocyte migration, cytokine release, microvascular thromboses, and cell death [3,4,5]. The proinflammatory phase of acute injury-associated systemic immune responses is often followed by an immunosuppressive phase [6]. During the pro-inflammatory phase, chemokines and cytokines are secreted by immune cells, and damage-associated molecular patterns (DAMPs) activate pattern recognition receptors (PRRs) [7]. Chronic injury is typically associated with metabolic rewiring, immune imbalance, and tissue remodeling [8, 9]. Most patients with organ injury receive temporary organ support or replacement therapies as there are no specific treatments to reverse or maintain individual organ functions [10]. Therefore, preventive and early supportive interventions are needed.
Epigenetic modifications regulate gene expression and developmental programs in the absence of changes in the gene sequence. Epigenetic modifications include histone modification, DNA methylation, chromosome remodeling, and regulation of transcription or translation by non-coding RNAs [11]. Histone acetylation and deacetylation have been extensively studied in recent years. Histones are intra-nuclear cationic proteins expressed in eukaryotic cells. They modulate gene expression by stabilizing chromatin structure; hence, alterations in histone patterns have been implicated in various diseases [12]. Histone acetylation was first described by Allfrey V in 1964 [13]. By regulating the acetylation of the N-terminal lysine residues of histones, histone acetyltransferases (HATs) and histone deacetylases (HDACs) determine chromatin structure and gene expression (Fig. 1A) [14]. Mounting evidence suggests that HDAC3 plays a key role in solid organ injury [15,16,17]. In this review article, we provide an overview of the current knowledge of the role of HDAC3 in the pathogenesis of solid organ injury, focusing on the possible underlying molecular mechanisms.

A Condensation and relaxation of chromatin due to histone deacetylation and acetylation, respectively. Histone acetylation levels are determined by the interplay between HATs and HDACs. Activation of HDACs leads to a net decrease of histone acetylation, chromatin condensation, and transcriptional repression. Activation of HATs results in a net increase of histone acetylation, chromatin relaxation, and transcriptional activation. B The chemical formula of histone acetylation and deacetylation. C Nuclear receptor co-repressor complexes containing HDAC3, GPS2, TBLX, and TBL1XR bind to nuclear receptors without ligands to induce transcriptional repression via histone deacetylation. D Nuclear receptor-mediated ligand binding inhibits the co-repressor complex and recruits co-activators, facilitating histone acetylation and gene transcription.
HATs and HDACs
Histones are critical components of nucleosomes. Post-translational modifications of histones affect chromatin structure and gene expression [18]. Histones are tightly coiled by DNA to form nucleosomes, contributing to the highly compact packaging of the eukaryotic genome [19]. The positively charged octamers of a linker histone (H1 or H5) and four highly basic histones (H3, H4, H2A, and H2B) interact with the negatively charged DNA via electrostatic bonds. H1 and H5 are responsible for the stabilization of chromosomes, promoting the formation of higher-order structures. Each histone octamer is tightly coiled by 147 base pairs of DNA [20]. These highly compact structures are dynamic and can be regulated by the binding of transcription factors to promoter sequences within the genome [21]. Epigenetic modifications can serve as markers to identify the different types of chromatin. For instance, heterochromatin is characterized by low acetylation levels, whereas euchromatin contains relatively high acetylation levels. Abnormal acetylation of histones may dysregulation gene expression, contributing to the occurrence of diseases [22].
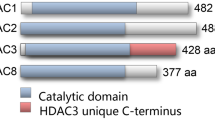
Histone acetylation and deacetylation levels are tightly regulated by the balance between the opposing activities of HATs and HDACs [23]. HATs are divided into three families: Gcn5-related acetyltransferases (GNATs); p300/CREB-binding proteins (CBP); and MOZ, Ybf2/Sas3, Sas2, and Tip60 (MYST)-related HATs. HATs catalyze lysine acetylation of histones by transferring the acetyl-CoA acetyl group to the ε-amino group of the internal lysine residue in the N-terminus. The addition of an acetyl group disrupts the electrostatic interaction between the DNA and histones. By neutralizing the positive lysine charge, histone acetylation alters chromatin structure and gene expression (Fig. 1B) [24, 25]. Deacetylation of histones is usually mediated by HDACs and promotes chromatin condensation and transcriptional repression [23]. In mammals, HDACs are divided into four categories: class I, II, III, and IV. Class I HDACs include HDAC1, HDAC2, HDAC3, and HDAC8. These HDACs are predominately found in the nucleus, but HDAC3 can translocate from the nucleus into the cytosol [26]. Class II HDACs include IIa HDACs (HDAC4, HDAC5, HDAC7, and HDAC9) and IIb HDACs (HDAC6 and HDAC10). Class IIa HDACs can shuttle between the nucleus and the cytosol. Class III HDACs are also known as sirtuins because of their homology to the yeast HDAC Sir2; this class includes SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7. Class IV HDACs only includes one member (HDAC11), which is predominantly expressed in the nucleus (Fig. 2). Class III HDACs are NAD-dependent enzymes, whereas the other HDACs are zinc-dependent enzymes [27, 28].

Class I, IIA, VI, and part of class III HDACs are mainly found in the nucleus. HDAC3, HDAC4, HDAC7, HDAC9, SIRT1, and SIRT2 shuffle between the nucleus and cytoplasm. Class IIB HDACs, including HDAC6 and HDAC10, are localized in the cytoplasm. Class III HDACs, including SIRT3, SIRT4, and SIRT5, are localized in mitochondria.
Structure and function of HDAC3
The unique structure of HDAC3
All class I HDAC members except HDAC8 share a similar structure, especially near the substrate-binding site [29]. Structural differences also exist between HDAC3 and other class I HDACs. HDAC3 possesses an aspartate residue at position 92, whereas HDAC1 and HDAC2 have a glutamate residue at this position at the outer rim of the cavity. Furthermore, HDAC1 and HDAC2 have a tyrosine residue at position 199, whereas HDAC3 has phenylalanine. Particularly, position 107 of HDAC3 is a tyrosine, while in the same position of HDAC1/2 is serine. Tyrosine 107 of HDAC3 leads to steric hindrance for binding to the foot pocket, precluding the binding of larger functional groups to inhibitors. Based on these differences, selective inhibitors against HDAC1, HDAC2, and HDAC3 can be designed [30, 31]. Hydrophobic amino acids at positions 13 and 29 of HDAC3 also differ from those in HADC1 and HDAC2, enabling the development of selective inhibitors targeting the foot pocket [32]. The biological function of HDAC3 requires nuclear receptor co-repressors, including silencing mediator of retinoic acid and thyroid hormone receptor (SMRT or NCoR2) and nuclear receptor co-repressor 1 (NCoR1) [33, 34]. Theoretically, the members involving 1, 2, 3, and 8 in the class I HDAC family may share similar functions and other members of the class I HDAC family will compensate when HDAC3 is knocked out effect. However, previous studies demonstrated that the expression of HDAC3 is not in accordance with HDAC1, HDAC2, and HDAC8 in the context of different pathological stimuli. Meanwhile, other members also display different biological functions from HDAC3 [35,36,37]. Therefore, we speculate that the other members in class I HDAC will not compensate when HDAC3 is absent based on the published articles. Of course, the expression of HDAC1, 2 and 8 should be also detected when HDAC3 was silenced in future studies.
Enzymatic activity and non-enzymatic functions of HDAC3
Nuclear receptors act as genetic switches regulating gene transcription by activating signal-dependent transcription factors. In turn, transcription factors integrate hormonal, metabolic, and environmental cues, recruiting various co-repressors and co-activators to specific genomic sequences [38]. HDAC3-containing nuclear receptor co-repressor complexes, including NCoR and SMRT, bind to ligand-free nuclear receptors, which directly repress gene expression. NCoR and SMRT complexes contain WD40 repeat-containing proteins, such as TBL1XR1 and TBL1X (Fig. 1C). These complexes recruit the 19S proteasome and the ubiquitylation machinery to histones [39]. G-protein pathway suppressor 2 (GPS2) is another core element of NCoR and SMRT complexes [40]; however, the role of GPS2 remains unclear. Noteworthily, under certain circumstances, HDAC3 indirectly activates gene expression. By contrast, nuclear receptor-mediated ligand binding inactivates the co-repressor complex and recruits co-activators, thereby facilitating gene transcription via histone acetylation [41] (Fig. 1D).
Enzymatic activity
The catalytic function of HDAC3 requires the physical interaction between HDAC3 and the deacetylase-activating domain (DAD) of NCoR and SMRT proteins. Crystal structure analysis of these complexes unveiled that abundant protein-protein interactions between the N-terminus of HDAC3 and the DAD of SMRT. Inositol tetraphosphate (Ins[1,4,5,6]P4 or IP4) has been identified, acting as “intermolecular glue” enhancing the interaction between HDAC3 and SMRT DAD via salt bridges and hydrogen bonds [42]. Intriguingly, once HDAC3 is dissociated from the NCoR or SMRT complex, it becomes unstable and is sequestered into a TCP1-ring complex in the cytoplasm [43]. TCP1 promotes HDAC3 folding in the cytoplasm, facilitating the formation of NCoR or SMRT complex containing an active HDAC3 enzyme in an ATP-dependent manner [44].
Non-enzymatic activity
In addition to its enzymatic functions, HDAC3 also displays non-enzymatic activities. Point mutations in Y298F, one of the active sites of HDAC3, disrupt its deacetylase activity. In the livers of HDAC3-deficient mice, the mutant HDAC3 can partially rescue hepatosteatosis and inhibit the expression of lipogenic genes, suggesting that HDAC3 has non-enzymatic functions [44]. Moreover, global HDAC3 knockout may lead to embryonic lethality because of gastrulation defects [45]. However, mutations in the DAD of SMRT and NCoR did not affect the Mendelian ratios of offspring mice, although the mice showed little HDAC3 enzymatic activity [45, 46]. Indeed, approximately 10% of enzymes with inactivating mutations in their active site are conserved in mammals, further supporting their non-catalytic functions [47]. Therefore, the non-enzymatic functions of HDAC3 should be taken into account in the development of HDAC3 inhibitors.
Physiological functions of HDAC3
Several genetically engineered mice with cell type-specific deletion of the Hdac3 gene have been developed recently to investigate the role of HDAC3. Notably, HDAC3 has been shown to regulate metabolism by increasing fatty acid oxidation and enhancing circadian histone deacetylation [47, 48]. HDAC3 also inhibits white adipose tissue metabolism by enhancing the futile cycle of fatty acid synthesis and oxidation and by decreasing acetylation in the enhancers of Ucp1 and Ppar-γ [48]. In addition, HDAC3 is essential for the development of vital organs. For example, mice with lung endodermal epithelium-specific Hdac3 knockout displayed lethality 2 to 10 days after birth due to defects in lung sacculation and early alveologenesis [49]. Similarly, cardiac progenitor cell-specific Hdac3 knockout led to ventricular septal defects and underdevelopment of ventricular walls, causing embryonic lethality [46]. Apart from lung and heart development, HDAC3 is also essential for the development and remodeling of bones [50].
Moreover, HDAC3 contributes to the maintenance of intestinal homeostasis and host defense. Notably, HDAC3 loss can cause inflammation and intestinal damage [50]. Furthermore, HDAC3-deficiency in neural progenitor cells impaired cortical lamination and neuronal migration, resulting in death within 16 h after birth. The critical role of HDAC3 in neuronal cell fate and function may be associated with HDAC3-mediated expression of T-box brain protein 1 [51]. Based on these findings, we conclude that HDAC3 expression and function are essential for multiple aspects of mammalian physiology and homeostasis (Fig. 3). The role of HDAC3 in organ injury is described below (Table 1).

Under physiological condition, HDAC3 is mainly responsible for the development and homeostasis of liver, heart, brain, lung, bone, pancreas, intestine, and adipocyte. However, the abnormal expression of HDAC3 also contributes to organ injury including heart, brain, pancreas, kidney, lung, and liver.
The emerging roles of HDAC3 in solid organ injury
Brain
Ischemic stroke is a potentially deadly cardiovascular disease causing significant morbidity and mortality worldwide. Ischemic stroke is usually triggered by the thrombus in the cerebral vasculature. Persistent occlusion in cerebral vasculature may hinder the supply of oxygen and glucose in the local brain tissue, eventually causing neuroinflammation, neuronal cell death, and secondary tissue injury during cerebral hypoperfusion and reperfusion [52, 53]. Type-1 interferons (IFNs) are pleiotropic cytokines regulating the expression of proinflammatory genes and orchestrating innate immune responses [54]. Hence, targeting IFNs or their upstream regulators may help prevent occlusion-induced brain injury [55, 56]. The cyclic GMP-AMP (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) pathway is a key regulator of the IFN pathway and innate immunity in response to double-stranded DNA (dsDNA) [57]. Microglia are innate immune cells residing in the central nervous system and serve as the principal effector cells contributing to neuroinflammation and brain injury caused by IRI. Conditional knockout of cGAS in microglia significantly relieved cerebral IRI. Mice with microglia-specific deletion of Hdac3 displayed low expression of cGAS at the mRNA and protein levels, as well as decreased levels of STING and IFN-γ. Mechanistically, HDAC3 deacetylates p65 at K122 and promotes the nuclear accumulation of p65, which regulates the transcription of cGAS [37]. HDAC3 inhibition using RGFP966 dampened the activation of melanoma 2 (AIM2) inflammasome in microglia, preventing ischemic brain injury [120]. As all class I HDACs have a zinc ion in the active site, small molecule inhibitors typically possess the same zinc-binding group. In addition, most HDAC inhibitors possess a linker connecting the zinc-binding group to a cap** group to mimic the lysine alkyl side chain. The most well-known zinc-binding group involves o-aminoanilides and hydroxamic acids. Entinostat (MS-275) and other selective class I inhibitors are based on o-aminoanilides. The selective HDAC3 inhibitor RGFP966 was developed through the modification of o-aminoanilides. PD106 and BRD3308 are also selective HDAC3 inhibitors used for the treatment of Friedreich’s ataxia, diabetes mellitus, and HIV infection [121]. Considering the potential of the o-aminoanilide scaffold in the development of selective HDAC3 inhibitors, many derivatives were synthesized based on this scaffold. In addition, some natural compounds can also regulate the expression or activity of HDAC3, exerting protective effects in various diseases in animal and cell models. For instance, juglanin prevents high-fat diet-induced renal injury by blocking the nuclear translocation of HDAC3 and NF-κB and thereby inhibiting inflammation and dyslipidemia [122]. miRNAs also play critical roles in nerve injury by targeting HDAC3 [123, 124]. The most common HDAC3 inhibitors and their roles in solid organ injury are summarized in Table 2. Most of the synthesized HDAC3 inhibitors lack specificity, as they partly inhibit other HDACs because of the high structural similarity of HDACs. Nearly all selective HDAC3 inhibitors are o-aminoanilide derivatives, the selectivity of which is mainly accessed through testing their IC50s [125]. However, this test method is not accurate. Alternatively, the Ki value of the o-aminoanilides can be determined by testing the kon and koff values of the inhibitors directly.
To build up a better picture of the regulatory mechanism of HDAC3, the activation mechanism of HDAC3 should also be paid particular attention. To our knowledge, there are mainly 3 aspects to promote the expression or the activity of HDAC3 at present. On the one hand, HDAC3 could be transcriptionally activated by certain transcription factors including Smad3 including Smad3 through directly binding its promoter [89]. On the other hand, HDAC3 activity could be enhanced via deubiquitinating. For example, HDAC3 activity could be enhanced by ubiquitin-specific protease 38 (USP38) via deubiquitinating. Notably, USP38 knockdown and overexpression could not change the protein level of HDAC3 [126]. In addition, PIWIL2 can also interact with HDAC3, giving rise to the stabilization of HDAC3 from ubiquitin-mediated degradation via competitive relation with E3 ubiquitin ligase Siah2. Meanwhile, PIWIL2 facilitated the interaction between CK2α and HDAC3, thus enhancing HDAC3 activity (Doi: 10.1038/s41419-018-0462-8). Last but not least, the microbiota-derived metabolite [inositol-1,4,5-trisphosphate (InsP3)] could promote epithelial repair by promoting HDAC3 activity, but the detailed mechanism remains unclear [127].
Clinical value of HDAC3
Till now, a great many clinical studies have also established the clinical position of HDAC3 in solid organ injury. For instance, compared with normal-weight women, obese individuals owned reduced levels of HDAC1, HDAC3, as well as HDAC9 in adipose tissues. Meanwhile, the mRNA levels and activity of these HDACs displayed an inverse correlation with inflammatory markers, obesity indices, and insulin levels, indicating that these HDACs have adverse effects on the development of obesity and diabetes mellitus [128]. One Chinese case-control study also demonstrated that the single nucleotide polymorphism of HDAC3 including rs11741808, rs2547547, and rs2547547 polymorphism was related to type 2 diabetes mellitus [129]. And combined HDAC3 and HDAC9 genes with diabetes mellitus worsen atherosclerosis and resulted in stroke [130, 131]. These clinical studies suggested that the genetic change of HDAC3 is closely associated with the development of diabetes mellitus and cerebrovascular disease. However, few clinical studies have reported the roles of HDAC3 in other types of solid organ injury. Before we reach clinical attempts, we might have to obtain more clinical data on HDAC3 to comprehensively harness the knowledge to benefit humans.
Future perspective and conclusion
HDAC3 is a unique and important member of the HDAC family. The catalytic activity of HDAC3 mainly depends on the integrity of nuclear receptor co-repressor complexes. In this review, we summarized the enzymatic and non-enzymatic functions of HDCA3. The non-enzymatic functions of HDAC3 should be taken into consideration when develo** new HDAC3-targeting strategies. We also briefly discussed the role of HDAC3 in the integration of various signals from the environment to regulate cellular fate, development, metabolism, and energy homeostasis. We also described the role of HDAC3 in the injury of solid organs, including the brain, heart, kidneys, liver, lungs, and pancreas. Although HDAC3 is beneficial under physiological conditions, pathological HDAC3 upregulation is detrimental in the pathogenesis of solid organ injury.
No human disease-causing mutations have been identified in the HDAC3 gene thus far, possible because germline mutations in HDAC3 may be may embryonically lethal in humans. However, numerous single-nucleotide polymorphisms (SNPs) in HDAC3 genes have been reported [130, 132]. We speculate that some SNPs may lie within enhancers and that these SNPs may hinder the recruitment of HDAC3 because of impaired binding of nuclear receptors or transcription factors, dysregulating HDAC3 expression. Another glaring question is how to overcome the high structural similarity of zinc-dependent HDAC isoenzymes to develop specific HDAC3 inhibitors rather than pan-HDAC inhibitors. The biological functions of HDAC3 should also be further explored in future studies.
Facts
Epigenetic modifications could regulate gene activity, as well as the development of an organism without changing gene sequence.
The reversible acetylation on the N-terminal lysine residues of histone proteins by histone HATs and histone deacetylases HDACs synergistically determine chromatin structure and gene expression.
HDAC3 is closely correlated with acute/chronic injury in the brain, heart, kidney, liver, lung, and pancreas.
Open questions
What is the physiological function of HDAC3 in development and homeostasis?
What are the precise molecular mechanisms of HDAC3 in solid organ injury?
What are the current situations and challenges in develo** novel drugs targeting HDAC3 for solid organ injury?
Data availability
No applicable resources were generated or analyzed during this study.
References
Dupont A, Rauch A, Staessens S, Moussa M, Rosa M, Corseaux D, et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscl Thrombosis Vascular Biol. 2021;41:1760–73.
Zhou H, Ma Q, Zhu P, Ren J, Reiter R, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 2018;64:e12471.
Cen M, Ouyang W, Zhang W, Yang L, Lin X, Dai M, et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 2021;41:101936.
Peukert K, Fox M, Schulz S, Feuerborn C, Frede S, Putensen C, et al. Inhibition of caspase-1 with tetracycline ameliorates acute lung injury. Am J Respirat Critical Care Med. 2021;204:53–63.
Tsai K, Chou W, Cheng H, Huang Y, Chang M, Chan S. Anti-IL-20 antibody protects against ischemia/reperfusion-impaired myocardial function through modulation of oxidative injuries, inflammation and cardiac remodeling. Antioxidants. 2021;10:275.
Chakraborty S, Karasu E, Huber-Lang M. Complement after trauma: suturing innate and adaptive immunity. Front Immunol. 2018;9:2050.
Lenz A, Franklin G, Cheadle W. Systemic inflammation after trauma. Injury. 2007;38:1336–45.
Ritchie R, Abel E. Basic mechanisms of diabetic heart disease. Circulation Res. 2020;126:1501–25.
Alaeddine L, Harb F, Hamza M, Dia B, Mogharbil N, Azar N, et al. Pharmacological regulation of cytochrome P450 metabolites of arachidonic acid attenuates cardiac injury in diabetic rats: the role of cytochromes P450 metabolites in diabetic cardiomyopathy. Transl Res. 2021;S1931-5244:00073–6
Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015;6:e1975.
Sodum N, Kumar G, Bojja S, Kumar N, Rao C. Epigenetics in NAFLD/NASH: targets and therapy. Pharmacol Res. 2021;167:105484
Ghoneim M, Fuchs H, Musselman C. Histone tail conformations: a fuzzy affair with DNA. Trends Biochem Sci. 2021;46:564–78
Allfrey V, Faulkner R, Mirsky AS. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc Natl Acad Sci USA. 1964;51:786–94.
Harrison I, Dexter D. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol. Therapeut. 2013;140:34–52.
Qiu Z, Ming H, Lei S, Zhou B, Zhao B, Yu Y, et al. Roles of HDAC3-orchestrated circadian clock gene oscillations in diabetic rats following myocardial ischaemia/reperfusion injury. Cell Death Dis. 2021;12:43.
Ghiboub M, Zhao J, Li Yim A, Schilderink R, Verseijden C, van Hamersveld P, et al. HDAC3 mediates the inflammatory response and LPS tolerance in human monocytes and macrophages. Front Immunol. 2020;11:550769.
Lin W, Zhang Q, Liu L, Yin S, Liu Z, Cao W. Klotho restoration via acetylation of peroxisome proliferation-activated receptor γ reduces the progression of chronic kidney disease. Kidney Int. 2017;92:669–79.
Xu H, Wu M, Ma X, Huang W, Xu Y. Function and mechanism of novel histone posttranslational modifications in health and disease. BioMed Res Int. 2021;2021:6635225.
Campos E, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99.
Stevens K, Swadling J, Hocher A, Bang C, Gribaldo S, Schmitz R, et al. Histone variants in archaea and the evolution of combinatorial chromatin complexity. Proc Natl Acad Sci USA. 2020;117:33384–95.
Tolsma T, Hansen J. Post-translational modifications and chromatin dynamics. Essays Biochem. 2019;63:89–96.
Neganova M, Klochkov S, Aleksandrova Y, Aliev G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin Cancer Biol. 2020;S1044-579X:30176-0.
Zhou Y, Peng J, Jiang S. Role of histone acetyltransferases and histone deacetylases in adipocyte differentiation and adipogenesis. Eur J Cell Biol. 2014;93:170–7.
Kumar V, Thakur J, Prasad M. Histone acetylation dynamics regulating plant development and stress responses. Cell Mol Life Sci. 2021;78:4467–86.
Gomathi K, Akshaya N, Srinaath N, Rohini M, Selvamurugan N. Histone acetyl transferases and their epigenetic impact on bone remodeling. Int J Biol Macromolecules. 2021;170:326–35.
Zhang K, Liu Z, Yao Y, Qiu Y, Li F, Chen D, et al. Structure-based design of a selective class I histone deacetylase (HDAC) near-Infrared (NIR) probe for epigenetic regulation detection in triple-negative breast cancer (TNBC). J Med Chem. 2021.64:4020–33.
Lee J, Bollschweiler D, Schäfer T, Huber R. Structural basis for the regulation of nucleosome recognition and HDAC activity by histone deacetylase assemblies. Sci Adv. 2021;7:eabd4413.
Chen X, He Y, Fu W, Sahebkar A, Tan Y, Xu S, et al. Histone Deacetylases (HDACs) and atherosclerosis: a mechanistic and pharmacological review. Front Cell Dev Biol. 2020;8:581015.
Maolanon A, Madsen A, Olsen C. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem Biol. 2016;23:759–68.
Methot J, Chakravarty P, Chenard M, Close J, Cruz J, Dahlberg W, et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg Med Chem Lett. 2008;18:973–8.
Wagner FF, Weïwer M, Steinbacher S, Schomburg A, Reinemer P, Gale JP, et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorganic Med Chem. 2016;24:4008–15.
Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, Jong RD, et al. Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Medicinal Chem Lett. 2010;20:3142–5.
Li J, Wang J, Wang J, Nawaz Z, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3 [In Process Citation]. EMBO J. 2000;19:4342–50.
Li J. Both corepressor proteins SMRT and N‐CoR exist in large protein complexes containing HDAC3. EMBO J. 2014;19:4342–50.
Zhang G, Zhou Z, Guo J, Gu H, Su M, Yu B, et al. Histone deacetylase 3 in hippocampus contributes to memory impairment after chronic constriction injury of sciatic nerve in mice. Pain. 2021;162:382–95.
Janczura K, Volmar C, Sartor G, Rao S, Ricciardi N, Lambert G, et al. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc Natl Acad Sci USA. 2018;115:E11148–E11157.
Liao Y, Cheng J, Kong X, Li S, Li X, Zhang M, et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. 2020;10:9644–62.
Everett LJ, Lazar MA. Cell-specific integration of nuclear receptor function at the genome. Wiley Interdiscip Rev Syst Biol Med. 2013;5:615–29.
Yoon H, Chan D, Huang Z, Li J, Fondell J, Qin J, et al. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003;22:1336–46.
Zhang J, Kalkum M, Chait B, Roeder R. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–23.
Emmett M, Lazar M. Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol. 2019;20:102–15.
Watson P, Fairall L, Santos G, Schwabe J. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–40.
Guenther M, Yu J, Kao G, Yen T, Lazar M. Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev. 2002;16:3130–5.
Sun Z, Feng D, Fang B, Mullican S, You S, Lim H, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–82.
Montgomery RL, Potthoff MJ, Haberland M, Qi X, Olson EN. Maintenance of cardiac energy by histone deacetylase 3 metabolism in mice. J Clin Investig. 2008;118:3588–97.
You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–87.
Adrain C, Freeman M. New lives for old: evolution of pseudoenzyme function illustrated by iRhoms. Nat Rev Mol Cell Biol. 2012;13:489–98.
Ferrari A, Longo R, Fiorino E, Silva R, Mitro N, Cermenati G, et al. HDAC3 is a molecular brake of the metabolic switch supporting white adipose tissue browning. Nat Commun. 2017;8:93.
Wang Y, Frank D, Morley M, Zhou S, Wang X, Lu M, et al. HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17-92 and TGF-β signaling regulation. Dev Cell. 2016;36:303–15.
Carpio L, Bradley E, McGee-Lawrence M, Weivoda M, Poston D, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9:ra79.
Norwood J, Franklin J, Sharma D, D’Mello S. Histone deacetylase 3 is necessary for proper brain development. J Biol Chem. 2014;289:34569–82.
Duan J, Gao S, Tu S, Lenahan C, Shao A, Sheng J. Pathophysiology and therapeutic Potential of NADPH oxidases in ischemic stroke-induced oxidative stress. Oxid Med Cell Longev. 2021;2021:6631805.
Taskiran-Sag A, Yemisci M, Gursoy-Ozdemir Y, Erdener S, Karatas H, Yuce D, et al. Improving microcirculatory reperfusion reduces parenchymal oxygen radical formation and provides neuroprotection. Stroke. 2018;49:1267–75.
Minter M, Zhang M, Ates R, Taylor J, Crack P. Type-1 interferons contribute to oxygen glucose deprivation induced neuro-inflammation in BE(2)M17 human neuroblastoma cells. J. Neuroinflammation. 2014;11:43.
MZ A, CED A, CHYW B, KMB A, GA A, JG C, et al. Type-I interferon signalling through IFNAR1 plays a deleterious role in the outcome after stroke. Neurochemistry Int. 2017;108:472–80.
Li L, Qin J, Guo S, Zhang P, Gong J, Zhang X, et al. Attenuation of cerebral ischemic injury in interferon regulatory factor 3-deficient rat. J Neurochem. 2016;136:871–83.
Bai J, Liu F. The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes. 2019;68:1099–108.
Zhang M, Zhao Q, **a M, Chen J, Chen Y, Cao X, et al. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 2020;34:648–62.
**a M, Zhao Q, Zhang H, Chen Y, Yuan Z, Xu Y, et al. Proteomic analysis of HDAC3 selective inhibitor in the regulation of inflammatory response of primary microglia. Neural Plasticity. 2017;2017:6237351.
Shou J, Zhou L, Zhu S, Zhang X. Diabetes is an independent risk factor for stroke recurrence in stroke patients: a meta-analysis. J Stroke Cerebrovasc Dis. 2015;24:1961–8.
Zhao B, Yuan Q, Hou J, **a Z, Zhan L, Li M, et al. Inhibition of HDAC3 ameliorates cerebral ischemia reperfusion injury in diabetic mice in vivo and in vitro. J Diabetes Res. 2019;2019:8520856.
Selkoe D, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2010;35:870–80.
Yu L, Liu Y, ** Y, Cao X, Chen J, ** J, et al. Lentivirus-mediated HDAC3 inhibition attenuates oxidative stress in APPswe/PS1dE9 Mice. J Alzheimer’s Dis. 2018;61:1411–24.
Borrelli M, Turnquist H, Little S. Biologics and their delivery systems: trends in myocardial infarction. Adv Drug Delivery Rev. 2021;173:181–215
Liem D, Zhao P, Angelis E, Chan S, Zhang J, Wang G, et al. Cyclin-dependent kinase 2 signaling regulates myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;45:610–6.
Song K, Li L, Quan Q, Wei Y, Hu S. Inhibited histone deacetylase 3 ameliorates myocardial ischemia-reperfusion injury in a rat model by elevating microRNA-19a-3p and reducing cyclin-dependent kinase 2. IUBMB Life. 2020;72:2696–709.
Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:jem.20171274.
Mia M, Cibi D, Abdul Ghani S, Song W, Tee N, Ghosh S, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020;18:e3000941.
Mullican S, Gaddis C, Alenghat T, Nair M, Giacomin P, Everett L, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25:2480–8.
Yahagi K, Kolodgie FD, Lutter C, Mori H, Virmani R. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2017;37:191–204.
Hsieh P, Zhang L, Jain M. Coordination of cardiac rhythmic output and circadian metabolic regulation in the heart. Cell Mol Life Sci. 2018;75:403–16.
Ikeda R, Tsuchiya Y, Koike N, Umemura Y, Inokawa H, Ono R, et al. REV-ERBα and REV-ERBβ function as key factors regulating mammalian circadian output. Sci Rep. 2019;9:10171.
Huang S, Chen G, Sun J, Chen Y, Wang N, Dong Y, et al. Histone deacetylase 3 inhibition alleviates type 2 diabetes mellitus-induced endothelial dysfunction via Nrf2. Cell Commun Signal. 2021;19:35.
Zhou H, Ren J, Toan S, Mui D. Role of mitochondrial quality surveillance in myocardial infarction: from bench to bedside. Ageing Res Rev. 2021;66:101250.
Pellicori P, Platz E, Dauw J, Ter Maaten J, Martens P, Pivetta E, et al. Ultrasound imaging of congestion in heart failure: examinations beyond the heart. Eur J Heart Failure. 2020;23:703–12
Sharifi-Sanjani M, Shoushtari A, Quiroz M, Baust J, Sestito S, Mosher M, et al. Cardiac CD47 drives left ventricular heart failure through Ca2+-CaMKII-regulated induction of HDAC3. J Am Heart Assoc. 2014;3:e000670.
Sun Z, Singh N, Mullican S, Everett L, Li L, Yuan L, et al. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J Biol Chem. 2011;286:33301–9.
Na J, ** H, Wang X, Huang K, Sun S, Li Q, et al. The crosstalk of HDAC3, microRNA-18a and ADRB3 in the progression of heart failure. Cell Biosci. 2021;11:31.
Zhang M, Yang X, Zimmerman R, Wang Q, Ross M, Granger J, et al. CaMKII exacerbates heart failure progression by activating class I HDACs. J Mol Cell Cardiol. 2020;149:73–81.
Li B, Yu Y, Liu K, Zhang Y, Qi J. β-Hydroxybutyrate inhibits histone deacetylase 3 to promote claudin-5 generation and attenuate cardiac microvascular hyperpermeability in diabetes. Diabetologia. 2021;64:1–14.
Zeisberg M, Neilson E. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrology. 2010;21:1819–34.
Gifford C, Tang J, Costello A, Khakoo N, Nguyen T, Goldschmeding R, et al. Negative regulators of TGF-β1 signaling in renal fibrosis; pathological mechanisms and novel therapeutic opportunities. Clin Sci. 2021;135:275–303.
Liu G, Philp A, Corte T, Travis M, Schilter H, Hansbro N, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Therapeut. 2021;225:107839.
Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J Exp Med. 2020;217:e20190103.
Liu Y, Bi X, **ong J, Han W, **ao T, Xu X, et al. MicroRNA-34a promotes renal fibrosis by downregulation of Klotho in Tubular Epithelial Cells. Molecular therapy. J Am Soc Gene Ther. 2019;27:1051–65.
Doi S, Zou Y, Togao O, Pastor J, John G, Wang L, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–65.
Satoh M, Nagasu H, Morita Y, Yamaguchi T, Kanwar Y, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Ren Physiol. 2012;303:F1641–1651.
Chen F, Gao Q, Wei A, Chen X, Shi Y, Wang H, et al. Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. Cell Death Differ. 2021;28:1001–12.
Levine M, Wang Z, Bhatti T, Wang Y, Aufhauser D, McNeal S, et al. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am J Transplant. 2015;15:965–73.
Ahola A, Harjutsalo V, Forsblom C, Pouwer F, Groop P. Depression is associated with progression of diabetic nephropathy in type 1 diabetes. Diabetes Care. 2021;44:174–80.
Shan Q, Zheng G, Zhu A, Cao L, Lu J, Wu D, et al. Epigenetic modification of miR-10a regulates renal damage by targeting CREB1 in type 2 diabetes mellitus. Toxicol Appl Pharmacol. 2016;306:134–43.
Hu L, Wang B, Jiang Y, Zhu B, Wang C, Yu Q, et al. Risk factors for transfusion-related acute lung injury. Respiratory Care. 2021;66:1029–38.
Zhan X, Cui R, Geng X, Li J, Zhou Y, He L, et al. LPS-induced mitochondrial DNA synthesis and release facilitate RAD50-dependent acute lung injury. Signal Transduct Target Ther. 2021;6:103.
Pooladanda V, Thatikonda S, Bale S, Pattnaik B, Sigalapalli D, Bathini N, et al. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death Dis. 2019;10:81.
Leus N, van der Wouden P, van den Bosch T, Hooghiemstra W, Ourailidou M, Kistemaker L, et al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem. Pharmacol. 2016;108:58–74.
Nguyen H, Adlanmerini M, Hauck A, Lazar M. Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature. 2020;584:286–90.
Gu L, Sun H, Chen J. Histone deacetylases 3 deletion restrains PM2.5-induced mice lung injury by regulating NF-κB and TGF-β/Smad2/3 signaling pathways. Biomed Pharmacother. 2017;85:756–62.
Hu D, Jia X, Cui L, Liu J, Chen J, Wang Y, et al. Exposure to fine particulate matter promotes platelet activation and thrombosis via obesity-related inflammation. J Hazard Mater. 2021;413:125341.
Cheng W, Lu J, Wang B, Sun L, Zhu B, Zhou F, et al. Inhibition of inflammation-induced injury and cell migration by coelonin and militarine in PM-exposed human lung alveolar epithelial A549 cells. Eur J Pharmacol. 2021;896:173931.
Niu X, Jones T, BéruBé K, Chuang H, Sun J, Ho K. The oxidative capacity of indoor source combustion derived particulate matter and resulting respiratory toxicity. Sci Total Environ. 2020;767:144391.
Jia H, Liu Y, Guo D, He W, Zhao L, **a S. PM2.5-induced pulmonary inflammation via activating of the NLRP3/caspase-1 signaling pathway. Environ Toxicol. 2021;36:298–307.
Xu F, Xu A, Guo Y, Bai Q, Wu X, Ji S, et al. PM2.5 exposure induces alveolar epithelial cell apoptosis and causes emphysema through p53/Siva-1. Eur Rev Med Pharmacol Sci. 2020;24:3943–50.
Jayachandran M, Brunn G, Karnicki K, Miller R, Owen W, Miller V. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J Appl Physiol. 2007;102:429–33.
Ahmad M, Shapiro M. Preventing diabetes and atherosclerosis in the cardiometabolic syndrome. Curr Atherosclerosis Rep. 2021;23:16.
Kim Y, Shin J, Kyeong D, Cho S, Kim M, Lim H, et al. Ahnak deficiency attenuates high-fat diet-induced fatty liver in mice through FGF21 induction. Exp Mol Med. 2021;53:468–82.
Zhang J, Xu Z, Gu J, Jiang S, Liu Q, Zheng Y, et al. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am J Physiol Endocrinol Metab. 2018;315:E150–E162.
Whitt J, Woo V, Lee P, Moncivaiz J, Haberman Y, Denson L, et al. Disruption of epithelial HDAC3 in intestine prevents diet-induced obesity in mice. Gastroenterology. 2018;155:501–13.
Armour S, Remsberg J, Damle M, Sidoli S, Ho W, Li Z, et al. An HDAC3-PROX1 corepressor module acts on HNF4α to control hepatic triglycerides. Nat Commun. 2017;8:549.
Kaltezioti V, Foskolou I, Lavigne M, Ninou E, Tsampoula M, Fousteri M, et al. Prox1 inhibits neurite outgrowth during central nervous system development. Cell Mol Life Sci. 2020;78:3443–65
Cnop M, Welsh N, Jonas J-C, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic Β-cell death in type 1 and type 2 diabetes. Diabetes. 2005;54:S97–S107.
Standards of Medical Care in Diabetes-20192. Classification and diagnosis of diabetes. Diabetes Care. 2019;42:S13–S28.
Deng C, **ang Y, Tan T, Ren Z, Cao C, Huang G, et al. Altered peripheral B-lymphocyte subsets in type 1 diabetes and latent autoimmune diabetes in adults. Diabetes Care. 2016;39:434–40.
Hu Q, Che G, Yang Y, **e H, Tian J. microRNA-296-5p/Bcl-xlHistone deacetylase 3 aggravates type 1 diabetes mellitus by inhibiting lymphocyte apoptosis through the axis. Front Genet. 2020;11:536854.
Lundh M, Christensen DP, Nielsen M, Richardson SJ, Dahllf MS, Skovgaard T, et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia. 2012;55:2421–2431.
Lei L, Bai G, Wang X, Liu S, **a J, Wu S, et al. Histone deacetylase 3-selective inhibitor RGFP966 ameliorates impaired glucose tolerance through β-cell protection. Toxicol Appl Pharmacol. 2020;406:115189.
Zhang Y, Li M, Wang Y, Liu X, Zhou L, Zhang C, et al. Histone deacetylase inhibition by MS-275 potentiates glucose-stimulated insulin secretion without affecting glucose oxidation. Life Sci. 2020;257:118073.
Lee SJ, Choi SE, Han BL, Song MW, Lee KW. A class I histone deacetylase inhibitor attenuates insulin resistance and inflammation in palmitate-treated C2C12 myotubes and muscle of HF/HFr diet mice. Front Pharmacol. 2020;11:601448.
Wagner F, Lundh M, Kaya T, McCarren P, Zhang Y, Chattopadhyay S, et al. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in β-cell protection. ACS Chem Biol. 2016;11:363–74.
Cao F, Zwinderman M, Dekker F. The process and strategy for develo** selective histone deacetylase 3 inhibitors. Molecules. 2018;23:551.
Sarkar R, Banerjee S, Amin S, Adhikari N, Jha T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: a review. Eur J Med Chem. 2020;192:112171.
Li Q, Ge C, Tan J, Sun Y, Kuang Q, Dai X, et al. Juglanin protects against high fat diet-induced renal injury by suppressing inflammation and dyslipidemia via regulating NF-κB/HDAC3 signaling. Int Immunopharmacol. 2021;95:107340.
Zhou Q, Feng X, Ye F, Lei F, Jia X, Feng D. miR-27a promotion resulting from silencing of HDAC3 facilitates the recovery of spinal cord injury by inhibiting PAK6 expression in rats. Life Sci. 2020;260:118098.
Zhao H, Li G, Zhang S, Li F, Wang R, Tao Z, et al. Inhibition of histone deacetylase 3 by MiR-494 alleviates neuronal loss and improves neurological recovery in experimental stroke. J Cereb Blood Flow Metab. 2019;39:2392–405.
Chou C, Herman D, Gottesfeld J. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–9.
Zhan W, Liao X, Liu J, Tian T, Yu L, Li R. USP38 regulates the stemness and chemoresistance of human colorectal cancer via regulation of HDAC3. Oncogenesis. 2020;9:48.
Wu S, Hashimoto-Hill S, Woo V, Eshleman E, Whitt J, Engleman L, et al. Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature. 2020;586:108–12.
Jannat Ali Pour N, Meshkani R, Toolabi K, Mohassel Azadi S, Zand S, Emamgholipour S. Adipose tissue mRNA expression of HDAC1, HDAC3 and HDAC9 in obese women in relation to obesity indices and insulin resistance. Mol Biol Rep. 2020;47:3459–68.
Zeng Z, Liao R, Yao Z, Zhou W, Ye P, Zheng X, et al. Three single nucleotide variants of the HDAC gene are associated with type 2 diabetes mellitus in a Chinese population: a community-based case-control study. Gene. 2014;533:427–33.
Chiou H, Bai C, Lien L, Hu C, Jeng J, Tang S, et al. Interactive Effects of a Combination of the HDAC3 and HDAC9 Genes with Diabetes Mellitus on the Risk of Ischemic Stroke. Thrombosis Haemost. 2021;121:396–404.
Sathishkumar C, Prabu P, Balakumar M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenet. 2016;8:1–12.
Ciuculete D, Boström A, Voisin S, Philipps H, Titova O, Bandstein M, et al. A methylome-wide mQTL analysis reveals associations of methylation sites with GAD1 and HDAC3 SNPs and a general psychiatric risk score. Transl Psychiatry. 2017;7:e1002.
Yang X, Wu Q, Zhang L, Feng L. Inhibition of Histone Deacetylase 3 (HDAC3) mediates ischemic preconditioning and protects cortical neurons against ischemia in rats. Front Mol Neurosci. 2016;9:131.
Lee S, Choi S, Lee H, Song M, Kim Y, Jeong J, et al. A class I histone deacetylase inhibitor attenuates insulin resistance and inflammation in palmitate-treated C2C12 myotubes and muscle of HF/HFr diet mice. Front Pharmacol. 2020;11:601448.
Joshi A, Barabutis N, Birmpas C, Dimitropoulou C, Thangjam G, Cherian-Shaw M, et al. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1410–1419.
Qiu Z, Ming H, Zhang Y, Yu Y, Lei S, ** rats by regulating p38 MAPK signaling pathways and epigenetic modification. Brain Res. 2020;1745:146932.
Stanfield B, Purves T, Palmer S, Sullenger B, Welty-Wolf K, Haines K, et al. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PLoS ONE. 2021;16:e0245169.
Dirice E, Ng R, Martinez R, Hu J, Wagner F, Holson E, et al. Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J Biol Chem. 2017;292:17598–608.
Demyanenko S, Nikul V, Uzdensky A. The neuroprotective effect of the HDAC2/3 inhibitor MI192 on the penumbra after photothrombotic stroke in the mouse brain. Mol Neurobiol. 2020;57:239–48.
Shi X, Liu Y, Zhang D, **ao D. Valproic acid attenuates sepsis-induced myocardial dysfunction in rats by accelerating autophagy through the PTEN/AKT/mTOR pathway. Life Sci. 2019;232:116613.
Chen X, Wang H, Zhou M, Li X, Fang Z, Gao H, et al. Alproic acid attenuates traumatic brain injury-induced inflammation: involvement of autophagy and the Nrf2/ARE signaling pathway. Front Mol Neurosci. 2018;11:117.
Li J, Ge C, Xu M, Wang W, Yu R, Fan C, et al. Betaine recovers hypothalamic neural injury by inhibiting astrogliosis and inflammation in fructose-fed rats. Mol Nutr Food Res. 2015;59:189–202.
Meng Q, Yang G, Yang Y, Ding F, Hu F. Protective effects of histone deacetylase inhibition by Scriptaid on brain injury in neonatal rat models of cerebral ischemia and hypoxia. Int J Clin Exp Pathol. 2020;13:179–91.
Wen Q, Lau N, Weng H, Ye P, Du S, Li C, et al. Chrysophanol exerts anti-inflammatory activity by targeting histone deacetylase 3 through the high mobility group protein 1-nuclear transcription factor-kappa B signaling pathway in vivo and in vitro. Front Bioeng Biotechnol. 2020;8:623866.
Lai N, Wu D, Liang T, Pan P, Yuan G, Li X, et al. Systemic exosomal miR-193b-3p delivery attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage in mice. J Neuroinflammation. 2020;17:74.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81800343, 81770095, 81700093).
Author information
Authors and Affiliations
Contributions
LN was responsible for conceiving and designing the review and writing the manuscript. XR was in charge of conceiving and designing some portions of the review and writing the manuscript. WB reviewed and edited the manuscript and served as the corresponding author. GQ was the principal investigator of this study and served as the corresponding author.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by B. Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ning, L., Rui, X., Bo, W. et al. The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis 12, 734 (2021). https://doi.org/10.1038/s41419-021-04019-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04019-6
- Springer Nature Limited