
Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is characterised by a dismal prognosis; nonetheless, limited studies have unveiled the mechanisms underlying HNSCC relapse.
Methods
Next-generation sequencing was performed to identify the somatic mutations in 188 matched samples, including primary tumours, tumour-adjacent tissues (TATs), pre- and post-operative plasma, saliva and peripheral blood lymphocytes (PBLs) from 27 patients. The evolutionary relationship between TATs and tumours were analysed. The dynamic changes of tumour- and TAT-specific mutations in liquid biopsies were monitored together with survival analysis.
Results
Alterations were detected in 27 out of 27 and 19 out of 26 tumours and TATs, respectively. TP53 was the most prevalently mutated gene in TATs. Some TATs shared mutations with primary tumours, while some other TATs were evolutionarily unrelated to tumours. Notably, TP53 mutations in TATs are stringently associated with premalignant transformation and are indicative of worse survival (hazard ratio = 14.01). TAT-specific mutations were also detected in pre- and/or post-operative liquid biopsies and were indicative of disease relapse.
Conclusions
TATs might undergo the processes of premalignant transformation, tumorigenesis and eventually relapse by either inheriting tumorigenic mutations from ancestral clones where the tumour originated or gaining private mutations independent of primary tumours. Detection of tumour- and/or TAT-specific genetic alterations in post-operative biopsies shows profound potential in prognostic use.
Similar content being viewed by others

Background
Head and neck cancer represents one of the most prevalent and lethal malignant tumour type worldwide [1,2,3]. Head and neck squamous cell carcinoma (HNSCC) accounts for >90% of head and neck cancers. There is evidence of a causal association between human papillomavirus (HPV) infection and oropharyngeal cancer [4]. In the USA and several western countries, ~60–80% of oropharyngeal cancers are related to HPV infection [5,6,7]. However, in the Chinese population, the prevalence of HPV-16 infection was found only in 7.0% of HNSCC cases [8, 9]. Smoking and sustained alcohol use were the highest risk factors for HPV-16-negative (HPV−) HNSCC, which has worse survival compared to HPV-16-positive (HPV+) HNSCC [9, 10].
HNSCC remains stagnated with a poor 5-year overall survival rate [11]. One important reason is that measurable residual disease (MRD) is relatively common [12]. Another reason is that second primary tumours (SPTs) often occur in the adjacent anatomical field [13]. The concept of field cancerisation is postulated as the main theory to explain the high frequency of SPTs; it was described by Slaughter et al. in 1953 when studying the presence of histologically abnormal tissue surrounding oral squamous cell carcinoma [14]. As explained by Braakhuis et al. [15], Tabor et al. [16] and others, multiple genetic alterations, including loss of heterozygosity (LOH), microsatellite alterations, chromosomal instability and mutations in the TP53 gene, have been detected in tumour-adjacent “normal tissue” and surgical margins to assess the presence of an altered field [16, 17]. Moreover, several studies have demonstrated risk factors, including cigarette and cannabis smoking, HPV infection, etc., that lead to cancer-associated genetic alterations in tumour-adjacent “macroscopically normal” tissue and surgical margins [14, 18,19,20,21,22,23,24,25]. Previous studies have reported detectable arm-level copy number changes such as 6p gain and 8p loss in HNSCC normal adjacent tissues and upregulated expression of pathways, including those involved in epithelial-to-mesenchymal transition in the normal adjacent tissues of several cancer types [26, 27]. As yet, however, little has been reported to elucidate their evolutionary relationship with primary tumours and evaluate their prognostic potential for monitoring disease progression and predicting survival. The clonality origin of the cancerised field and the occurrence of multiple tumours remain controversial, as some scholars support the monoclonal theory in which multiple tumours originate from a single cell with oncogenic genetic changes, while others suggest that multiple transforming events might give rise to genetically unrelated multiple tumours [23]. Besides, the relation between field cancerisation with HNSCC relapse awaits exploration. Moreover, most of these studies focussed on the pathologically normal tumour-adjacent tissues (TATs); little attention has been paid to the adjacent tissues that remained unresected in the commonly accepted surgical guidelines [28, 29], of which some of these tissues might not be pathologically normal.
Fuelled with cutting-edge bioinformatics tools including next-generation sequencing (NGS), several genetic biomarkers for monitoring cancer status have been developed. Among them, tumour DNA can be detected in the blood and other body fluids of patients with advanced cancer [30,31,32]. Tumour DNA that enters the circulation becomes circulating tumour DNA (ctDNA) and is capable of reflecting genetic alternations dynamically [33, 34]. Many studies have shown that ctDNA levels rise rapidly with disease progression and subsequently drop in association with successful treatment [35,36,37]. ctDNA can also be extracted from other body fluids like saliva (SctDNA). Previous studies by Wang et al., Shanmugam et al., Ahn et al. and others found that ctDNAs were highly detectable in the saliva and plasma of patients with HPV+ or HPV− HNSCC, serving as liquid biopsy with diagnostic and monitoring applications [32, 38,39,40]. Besides, Lv et al. reported detecting circulating Epstein–Barr virus DNA copy number had substantial power to predict patients’ survival, serving as an alternative marker to commonly used ctDNA [41]. However, there is little knowledge about the cell-free DNA (cfDNA) that originated from TATs. Currently, the well-accepted method is to detect ctDNA with the somatic mutations derived from primary tumours. As TATs have the potential to develop into malignant lesions, they might be able to predict the disease progression as well.
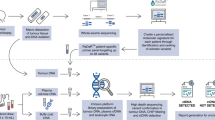
Herein, we reported a prospective study utilising targeted-capture NGS to profile the mutational features of samples including tumour tissues, TAT, pre- and post-surgical ctDNA and SctDNA from 27 HPV-16− HNSCC patients. A chart depicting the general workflow in this study was shown in Fig. S1. The result showed that the genetic mutations were highly detected in the saliva, plasma and TAT of HNSCC patients. By comparing the mutations in different sources of samples, we characterised two models of an evolutionary relationship between TATs and primary tumours, providing key information to understand HNSCC premalignant development, tumorigenesis and relapse. Finally, the prognostic value of TAT, ctDNA and SctDNA as biomarkers to monitor relapse in HNSCC were assessed.
Methods
The present study was a prospective study. All blood, saliva and adjacent tumour mucosa tissues were obtained from HNSCC patients at ** quality <30 or with paired-end reads bias were removed. The Genome Analysis Toolkit (https://www.broadinstitute.org/gatk/) and MuTect2 were employed to call somatic small insertions and deletions (indels) and single-nucleotide variants by filtering PBL sequencing data [40]. For liquid biopsies, mutations had also been detected in matched tumour tissues and/or TATs were kept. Phylogenic trees were illustrated using Phylip (version 3.698), by looking at the concordant mutations. Shared mutations were defined as trunk mutations, while private mutations were shown on branches. Clonality of mutations in tumours and TATs were inferred by looking at the variant allele fraction (VAF) ratio of mutations, calculated in the way as previously reported [49]. Specifically, the VAF ratio equals to dividing the VAF of each mutation by the maximum VAF in the same sample. In order to make sure the TATs were not contaminated by tumour tissues, we filtered out the somatic mutations with VAF <0.025. This was because only a low fraction of tumour cell DNA would be sequenced from the contaminating tissues, yielding a low VAF ratio of the corresponding mutations identified. Besides, some TAT-specific clonal mutations observed in the TATs were not observed in the matched tumour tissues, indicating that these mutations were not detected due to tumour sample contamination. Mutations with a VAF ratio >0.8 were determined as clonal, while the others were determined as subclonal [49].
Statistical analysis
Wilcoxon’s rank-sum test and Kruskal–Wallis test were used to compare differences between groups. Curves for disease-free survival (DFS) were constructed using the Kaplan–Meier method and compared using the log-rank test. Cox proportional hazards regression analysis was used to assess the impacts of ctDNA, SctDNA and adjacent mucosa mutations on DFS. All P values were based on two-sided tests and differences were considered significant at P ≤ 0.05. Statistical analysis was performed using the SPSS 17.0 and the GraphPad Prism 7 software.
Results
Patient characteristics
All patients were diagnosed with squamous cell carcinoma. Among them, 27 patients were determined to be HPV-16− by quantitative polymerase chain reaction and further subjected to downstream analyses. For each patient, the clinicopathological characteristics and follow-up data are given in Table S2. With a median follow-up time of 20.7 months (range 1.3–27.2 months), 8 of 27 patients (29.7%) developed recurrence after completing their treatment. A total of 188 prospectively collected pre- and post-operative plasma, saliva, adjacent mucosa, and PBL samples from the 27 HNSCC patients were analysed. Details of the patient enrolment, exclusion and sample treatment are presented in Fig. S2.
Mutations in primary tumours
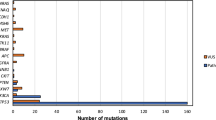
Tumour tissue DNA and matched PBL DNA from 27 HNSCC patients were sequenced to detect somatic mutations in each sample, with the method mentioned in a previous report [40], achieving a mean read depth in the range of 337×–2020× (without duplication) for tumour tissue DNA and 221×–308× (without duplication) for PBL DNA. We identified at least one somatic mutation in all of the primary tumours. The average number of somatic mutations identified in each primary tumour was 6.07 (range 1–13). A total of 164 somatic mutations, including SNVs and Indels, were identified in 91 genes, and 22 genes were recurrently mutated in multiple cases (Fig. 1). The most frequently mutated gene was TP53 (24/27, 88.89%). Other recurrently mutated genes included FAT1 (9/27, 33.33%); NOTCH1, TERT (5/27, 18.52%); NOTCH2 (4/27, 14.81%); EPHA3, EPHA5, KDR, PIK3CA and PTPRD (3/27, 11.11%). The tri-nucleotide changing patterns of SNVs in all samples were compared with the COSMIC (accessed in November 2020) categorised SBS, which was published by Alexandrov et al. from PCAWG studies [50], using the YAPSA software. The number of mutations attributed to each SBS was quantified and plotted in Figs. S3a and 3b. In our result, the highest numbers of SNVs were attributed to SBS20, which is related to DNA mismatch repair, SBS4, associated with tobacco smoking, and SBS7b/7c, owing to ultraviolet exposure.

Top panel, the mutational burden in each sample; right panel, the prevalence of mutations in each gene; bottom panel, from top to bottom, the source of the sample from the patients; pathological diagnosis of TAT sample from the respective patient (TAT unavailable in one case, labelled with grey); patients with or without relapse in follow-up; TNM stages of the patients. TAT tumour-adjacent tissue, pre-ctDNA pre-operative ctDNA, post-ctDNA post-operative ctDNA, pre-sctDNA pre-operative SctDNA, post-sctDNA post-operative SctDNA. Mutation types are labelled with different colours listed at the bottom left.
Mutations and histopathology in TAT
In order to profile the genomic alterations in adjacent mucosa, genomic variations in 26 TATs were identified as well. On average, adjacent mucosa tissues yielded a 731× (431×–1041×) sequencing depth of coverage. We identified at least one somatic SNV or Indel in 73.1% (19 of 26) of TATs. The average number of somatic SNVs and Indels identified in each TAT tissue was 3.54 (range 0–13). A total of 92 somatic SNVs and Indels were identified in 48 genes; seven genes were mutated in multiple TATs, including TP53 (12/26), NOTCH1 (6/26), FAT1 (4/26), CDKN2A (2/26), EPHA3 (2/26), MLH1 (2/26) and PIK3CA (2/26) (Figs. 1 and 2). We next tried to infer the histopathological status of these TATs. TATs were pathologically determined as a premalignant lesion (5/26, dysplasia) or negative surgical margins (21/26) as confirmed by two pathologists. Fisher’s exact test showed a significantly higher number of premalignant lesions in TP53mut TATs (P = 0.012, Table S3a), no matter if the same TP53 mutations could be found in the respective primary tumours. The mutant p53 protein expressed in TP53mut TATs was verified through immunohistochemistry analysis, which showed mild to strong mutant p53 expression in all of the TP53mut TATs (Table S3b and Fig. S3c).

a Somatic mutations in patients whose TATs shared at least one mutation with primary tumours. b Somatic mutations in patients whose TATs harboured no or private mutations to primary tumours. TAT tumour-adjacent tissue, pre-sctDNA pre-operative SctDNA, pre-ctDNA pre-operative ctDNA, post-sctDNA post-operative SctDNA, post-ctDNA post-operative ctDNA. Patients with a premalignant lesion diagnosed pathologically in TATs are labelled with star icons, while relapsed patients are labelled with hexagon icons. Tumour sites and TNM disease stages were labelled next to patients’ ID. Mutation types are labelled with different colours listed at the bottom right; TAT was not available in P25.
Evolutionary relationship between TATs and primary tumours
We then sought to answer if TATs and primary tumours were evolutionarily associated in HNSCC. Twenty-three SNVs and Indels were identified in both tissues of the same patients (Fig. 2), and in 30.8% (8/26) of cases, TATs shared at least one somatic SNV or Indel with the corresponding primary tumours. Nevertheless, 7 out of 26 patients harboured no mutations in their TATs, and 11 out of 26 patients only harboured private mutations in their TATs. Among these 11 patients, recurrent mutations in TATs were found in several canonical driver genes encompassing TP53, CHEK2, NOTCH1, ERBB4, FAT1, etc. Phylogenic analysis was then carried out to shed further light on the evolutionary relationship between TATs and primary tumours in 19 patients with mutations identified in their TATs (Fig. S4). The phylogenic trees of all the relapsed patients and patients with premalignant lesions are exhibited in Fig. 3a, wherein five out of eight patients’ TATs did not share common clonal origins with primary tumours. Relative numbers of tumour-private, TAT-private and shared SNVs and Indels in all patients with mutations identified in their TATs were summarised in Fig. 3b. Among all, 8/19 patients harboured at least one shared mutation between TATs and primary tumours, indicating a monoclonal origin in each pair of TAT and tumour, while multi-clonal origins were observed in the other 11 out of 19 patients (Figs. 3a and S5). The average number of tumour-private mutations was significantly higher than that of TAT-private mutations, while significantly more TAT-private mutations were observed than tumour-TAT shared mutations (Fig. 3c). In the group with monoclonal origins, five out of eight TATs shared the same TP53 mutations as in respective primary tumours. Other putative oncogenic trunk mutations shared in TAT–tumour pairs included mutations in MTOR, NOTCH1, MET, BRCA1 and CCNE1.

a Phylogenic tree constructed by looking at the concordant mutations in TATs and primary tumours. Arrows with different colours indicate the mutations that are detected in liquid biopsies: orange, pre-operative plasma; green, pre-operative saliva; purple, post-operative plasma; blue, post-operative saliva. TAT tumour-adjacent tissue; the number of mutations identified were labelled near each branch or trunk. Patients with a premalignant lesion diagnosed pathologically in TATs are labelled with star icons, while relapsed patients are labelled with hexagon icons. b Distribution of shared, TAT- and tumour-private mutations in each patient. c Average numbers of tumour-private, TAT-private and tumour-TAT shared mutations in all samples. Significant difference was difined as * (P < 0.05), ** (P < 0.01), or *** (P < 0.001) in a two-sided students’ t-test.
Notably, although TP53 might be altered both in primary tumour and TAT in the same patient, the mutations were not necessarily the same. For instance, in P03 (primary tumour: R248W and C135R vs. TAT: R248Q), P08 (primary tumour: I162Sfs*3 vs TAT: V97Afs*26, G266* and R273C), P16 (primary tumour: A159P and Y236* vs. TAT: R209*) and P19 (primary tumour: P250S, R249S and H214R vs. TAT: R248W, Y220C and Y205C), different TP53 alterations were observed between each pair of the primary tumour and TAT (Table S4). Besides, in certain patients, both shared and private TP53 mutations were observed in primary tumour–TAT pairs, such as P27 (primary tumour: I255del vs. TAT: I255del, Y220C and R273S). Details of TP53 alterations detected in all patients were exhibited in Table S4.
VAF/maximum VAF in tumours and TATs was calculated in order to estimate the likely cancer-cell fraction value and infer the clonality of each mutation in samples. The VAF ratio of all relapsed patients and patients with premalignant lesions were illustrated in Fig. 4, while Fig. S6 denotes the situations in all patients with mutations detected in their TATs. With a median VAF ratio of 0.93 (range 0.06–1), TP53 mutations were determined as clonal in 13 tumour tissues, and clonal TP53 mutations were observed in 9 TATs. Shared clonal mutations included MLH1 mutation in P06 and TP53 mutation in P27. Eight of 12 mutations detected post-surgery in plasma and/or saliva were clonal. Overall, the mean VAF ratio of mutations in CCNE1, KEAP1, FCGR2A, PTEN and TP53 were the highest in all primary samples, indicating that these mutations were early-stage clonal events during tumour progression (Fig. S6). In addition, TP53 also had the highest mean VAF ratio in premalignant TATs but not in pathologically normal TATs, suggesting that TP53 mutation might play a key role in promoting tissue premalignant transformation and tumourisation (Fig. S6).

VAF variant allelic frequency; mutations are labelled with different colours according to their clonal type in TATs and primary tumours, indicated at the bottom right. Mutations with VAF ratio >0.8 are defined as clonal, while other mutations are defined as subclonal. Arrows with different colours indicate the clones that are detected in post-operative plasma and saliva: purple, post-operative plasma; blue, post-operative saliva. Patients with premalignant lesion diagnosed pathologically in TATs are labelled with star icons, while relapsed patients are labelled with hexagon icons.
TP53 mutations in TAT are predictive of recurrence
In 21 patients whose TATs were determined as negative surgical margins, recurrence occurred ultimately in five patients, and mutations in TATs were detected in all of these patients. Fisher’s exact test showed no significant association between TAT–tumour evolutionary relationship with disease relapse (P = 0.28, Table S3c). Moreover, recurrence was eventually observed in three of the patients (P03, P06 and P27) whose TATs shared monoclonal origins with their primary tumours, while 4 (P09, P18, P19 and P26) out of 11 patients whose TATs harboured only private SNVs and/or Indels finally underwent relapse. Intriguingly, mutations in P6 who underwent relapse finally were observed identical to what was identified in the primary tumour, although the pathological tests determined this TAT as a negative surgical margin.
According to the COSMIC database and our previous studies [51], TP53 mutations are the most prevalent genetic alterations in HNSCCs, and they were detected in 50.0% of TATs in the present cohort. We thereby tested whether TP53 mutations in TAT could anticipate relapse. As a result, patients whose TATs harboured TP53 mutations had a greater tendency to relapse, and had significantly shorter DFS (hazard ratio (HR) 14.01; 95% confidence interval (CI), 2.48–79.28; P = 0.003; Fig. 5a). The Kaplan–Meier estimates of DFS were 11.3 months for the TP53 altered patients and 23.9 months for the wild-type TP53 patients. Multivariate Cox proportional hazard model yielded a P value of 0.01 when taking various clinicopathological characteristics into consideration (HR 6.76; 95% CI, 1.59-28.69; Table S5a). This indicates that these “tumour-free” margins determined by pathological means might still contain tumour cells or have the potential to develop into SPTs, which would probably lead to local recurrence. Besides, four patients with their TATs diagnosed pathologically as negative surgical margins finally underwent relapse, and mutations were detected in TATs of all these four patients, suggesting that predicting disease relapse solely based on TAT pathological diagnosis might not be sufficient and that TAT mutational features could provide extra valuable information for HNSCC prognosis.

a Disease-free survival according to the detection of TP53 mutation in adjacent mucosa sample. P value determined by log-rank test (n = 7 for TP53 mutation positive, n = 20 for TP53 mutation negative). b Mutation burdens in both post-surgical plasma and saliva fell sharply after resection compared with pre-surgery. P value determined by Wilcoxon’s rank-sum test. c Disease-free survival according to the detection of ctDNA in the post-operative plasma sample. P value determined by log-rank test (n = 6 for ctDNA positive, n = 21 for ctDNA negative). d Disease-free survival according to the detection of SctDNA in the post-operative saliva sample. P value determined by log-rank test (n = 7 for SctDNA positive, n = 20 for SctDNA negative).
Mutations in plasma and saliva
Targeted-capture sequencing of 27 HNSCC patients’ matched pre- and post-operative plasma ctDNA, saliva tumour DNA and matched PBL DNA was performed, achieving a mean sequencing coverage of 934× (370×–1716×) for plasma ctDNA, 1049× (259×–1865×) for saliva tumour DNA and 287× (221×–308×) for PBL DNA. The presence of ctDNA/SctDNA was evaluated by searching for the presence of previously identified SNVs and Indels identified in tumour tissue and/or TAT (Figs. 1 and 2). For pre-operative samples, mutations were identified in 70.4% (19/27) and 63.0% (17/27) of plasma and saliva samples, respectively. TP53 mutation was the most prevalent (68.4%, 13/19) alteration in plasma ctDNA, with FAT1, EPHA3, EPHA5 and PTPRD also being recurrently mutated (Fig. 2). The most frequently mutated gene in saliva SctDNA was also TP53 (88.2%, 15/17). GNAS, FAT1 and MLH1 were commonly mutated (14.3%, 2/14). For post-operative samples, nine mutations in plasma were identified in seven patients. Mutated genes included MLH1, TP53, AKT1 and FAT1. Twelve mutations in saliva were detected in 11 patients, where mutations in CDKN2A, TP53, MLH1, FAT1, NOTCH1, AKT1 and DHX9 were identified. In order to answer if the mutations detected in liquid biopsies were more likely to originate from tumours or TATs, the number of patients with tumour-private, TAT-private or tumour-TAT shared mutations detected in their pre-/post-operative liquid biopsies was calculated and plotted in Fig. S7, together with the average number of mutations detected in each type of biopsies at two time points. Notably, TAT-private mutations were detected in both plasma and saliva pre- and post-surgery, and all patients with TAT-private mutations detected in their post-operative plasma eventually underwent relapse.
Prognostic value of tumour-specific ctDNA and SctDNA in early post-operative plasma and saliva samples
Plasma and saliva samples were taken from 27 patients 1–2 weeks after surgery completion were analysed. Mutation burdens in both post-operative plasma and saliva clearly dropped after surgery (post vs. pre, P = 0.001 for ctDNA, P < 0.001 for SctDNA; Fig. 5b).
Six or seven out of eight relapsed patients had ctDNA detected in post-operative plasma and saliva accordingly. Of particular interest, in P03 who underwent recurrence eventually, only a TAT-private CDKN2A mutation was detected in post-operative plasma, which was not identified in the respective tumour. Similarly, private mutations only found in TATs were detected in post-operative saliva in four patients, among whom two underwent relapse ultimately. A common practice to use liquid biopsy for prognosis was to detect only tumour-specific mutations post surgery. With this method, ctDNA analysis of post-operative plasma predicted impending relapse with 62.5% sensitivity and 94.7% specificity (Table S5b). By comparison, tumour DNA analysis of post-operative saliva predicted relapse with 62.5% sensitivity and 89.5% specificity (Table S5b). In addition, post-operative ctDNA-positive patients had significantly shorter DFS than those with no ctDNA detected (hazard ratio (HR) 22.27; 95% confidence interval (CI), 3.593–138.1; P < 0.001; Fig. 5c). Similarly, SctDNA-positive patients also had considerably lower DFS than those who were SctDNA negative (HR 20.51; 95% CI, 3.359–125.3; P = 0.001; Fig. 5d). In multivariate analysis, Cox proportional hazard model yielded P values of 0.005 for both ctDNA and SctDNA when taking other clinicopathological characteristics into consideration (ctDNA HR 8.03; 95% CI, 1.89–34.13; SctDNA HR 7.85; 95% CI, 1.84–33.47; Table S5c and Table S5d, respectively). Notably, detecting TAT-specific mutations in addition to tumour-specific mutations in plasma and saliva increased the sensitivity of post-operative ctDNA from 62.5 to 75%, while the sensitivity of SctDNA increased from 62.5 to 87.5% in predicting relapse (Table S5e). Furthermore, patients with ctDNA or SctDNA post-surgical detection were confirmed to undergo relapse earlier compared with conventional clinical imaging (Fig. S8a, b).
Discussion
The detection of HNSCC recurrence following surgery is often delayed owing to the clinical challenges, which compromise clinical decision-making and impair patient management. A proportion of early-stage HNSCC patients are prone to relapse within a short time. Therefore, it is necessary to develop a surveillance test for early detection of MRD prior to the development of clinical symptoms. Besides, SPT at the tumour-adjacent fields, which appeared to be normal in traditional diagnostic tests, substantially increase the risk of recurrence.
Previously, Tada et al. exhibited that the EMT status of CTC might be a potential marker to predict disease progression [52]. Wang et al. and Hamana et al. demonstrated in cohorts of 47 HNSCC and 64 oral SCC patients that detectable post-operative ctDNA was a powerful surrogate to indicate patients’ relapse [53,54,55]. Besides, Wang et al. also showed that both ctDNA and SctDNA could improve the sensitivity of predicting relapse [53]. Here, we showed that TP53 mutations in TAT exhibited profound predictive power in the prognosis of HPV-16− HNSCC, allowing sensitive risk assessment of HNSCC recurrence. Notably, we showed that detecting TAT-specific mutations increased the sensitivity of ctDNA and SctDNA in monitoring MRD and SPT to predict relapse. These results rendered it promising to translate these biomarkers into clinical use for predicting HNSCC prognosis and guiding treatment decisions.
The concept of field cancerisation was proposed to demonstrate a proportion of cells with early genetic alterations but without histopathological change remaining in the TATs in HNSCC [56]. The clonal origin of the cancerised field and the occurrence of multiple tumours remain controversial, as some scholars support the monoclonal theory in which multiple tumours originate from a single cell with oncogenic genetic changes, while others suggest that multiple transforming events might give rise to genetically unrelated multiple tumours [23]. Besides, the relation between field cancerisation with HNSCC relapse awaits exploration. Previous studies utilised markers such as microsatellite instability and TP53 mutation to evaluate the clonality of multiple tumours in HNSCC and observed their polyclonal origins, which was consistent to our findings [23, 57, 58].
In our study, the evolutionary analysis showed that 8 out of 26 TATs shared monoclonal origins with tumours, where relapse occurred in three patients, and premalignant lesion in TAT was observed in one patient. In contrast, 11 out of 26 TATs harboured only private driver mutations, and four of these patients underwent recurrence eventually. In three relapsed patients only TAT-private mutations were detected in post-operative plasma or saliva, indicating that TATs might be evolutionarily independent of primary tumours during tumorigenesis. Of the TATs from these four patients, two was determined as premalignant lesions, while the other two were determined as negative surgical margins. Moreover, TAT-private mutations were detected in post-operative plasma and saliva, and all patients with TAT-private mutations detected in post-operative plasma eventually underwent recurrence. These results revealed that TAT might employ two different modi operandi to contribute to disease recurrence, as depicted in Fig. 6a. Some cells in TAT originated from the same ancestral clone as the tumour did; TAT cells might inherit the same driver alterations in the tumour that tend to occur in an early stage of tumorigenesis, and further obtain novel mutations triggering premalignant transformation and tumour progression, leading to disease relapse after resection (Fig. 6b). Private mutations might be accumulated in other cells that did not inherit the same driver mutations as in tumours, and later contribute to the development of an SPT that is evolutionarily independent of primary tumours, resulting in disease recurrence.

One ancestral clone carrying driver mutations such as TP53 mutation proliferate to produce mucosa tissue. Some of the offspring cells gain novel driver mutations to develop into primary tumours. After resection, a some cells in TAT originated from the same ancestral clone as tumours might inherit the same driver alterations in tumours that tend to occur in an early stage of tumorigenesis, and further obtain novel mutations triggering premalignant transformation and tumour progression, leading to disease relapse. b In some other cells that inherit no identical driver mutations as in primary tumour, private mutations might be accumulated, and later contribute to the development of a second primary tumour, which is evolutionarily independent of primary tumours, resulting in disease recurrence. Notably, cells sharing identical mutations with primary tumour might still exist in the TAT simultaneously, but they failed to develop into a new tumour.
Intriguingly, Fisher’s exact test exhibited no significant association between TAT-tumour clonality with TAT pathological status or disease recurrence, suggesting that a TAT-tumour monoclonal origin does not necessarily precede the TAT premalignant transformation and tumorigenesis, nor the disease relapse of HNSCC. On the other hand, one or two relapsed patients only harboured TAT-specific mutations in their post-operative plasma and saliva respectively, suggesting that relapse in these cases are more likely to be as a result of second independent tumours. Taken together, these results further solidify the conjecture that a proportion of recurrence in HNSCC is likely to result from premalignant transformation and tumorigenesis of TATs, which might not be evolutionarily associated with primary tumours and instead undergo tumorigenesis independently. However, some other studies showed identical genetic changes between multiple tumours in the head and neck [57, 59,60,61]. The possible reason behind the discrepancy between findings of these studies with ours might be the different markers used to define the clonality of tumours, given that the markers used in these studies, such as LOH patterns, cytogenetic features, etc., occur in a high frequency in HNSCC and are not specific enough to be distinguished when they occur independently [22, 57, 59,60,61].
We adopted 1021-panel targeted-capture sequencing, but not whole-exome sequencing or whole-genome sequencing, to infer the evolutionary relationship between tumour and adjacent mucosa; one possible drawback of this method is that we might miss some passenger mutations that are indeed shared by these two tissues. However, it is conceivably impossible to conclude that two tissues from the same patient are totally irrelevant during development; nonetheless, the shared alterations in passenger genes might not play essential roles in the process of tumour evolution. Previously, Wang et al. showed different sensitivities of liquid biopsies in detecting tumour mutations of HNSCC originating from different anatomical sites [12]. The conclusion from the present study was quite consistent with the previous one, in terms of that more tumour-derived mutations were detected in ctDNA of saliva among patients with tumours arising in the oral cavity, including the base of the tongue, tongue and buccal sites, while comparatively more mutations were detected in plasma for deeper tumours, such as those that arose in the larynx, etc. However, due to the modest sample size, no statistical significance was observed. We also looked at whether more mutations could be identified in tumours with higher disease stages, but no obvious pattern was scrutinised. Besides, no significant association could be observed between the detection of TP53 mutations in TAT and the detection of mutations in pre/post plasma or saliva with patients’ smoking history (Table S6a–e).
One major limitation of this study was the modest sample size, and as we focussed on the patients’ samples collected from real clinical practice, we might not be able to perform more wet experiments to elucidate mechanisms of genetic alterations contributing to disease relapse or TAT cancerisation. The limited sample size also led to insufficient amounts of identified Indels for conducting Indel signature analyses. Besides, even though we found a trend that alterations in pre-operative plasma were more likely to be detected in patients with higher TNM stages of disease than with lower stages, only near-borderline significance (P = 0.07) (Fig. S6e) could be observed, possibly due to the modest sample size as well. Notably, this result was consistent with the findings of previous studies, which suggested that a higher level of cfDNA in pre-operative plasma was observed in patients with higher stages of the disease [62, 63]. More investigations with larger sample sizes and more approaches to explain the cause and consequence relationship in these processes are needed. In the present study, the detection rates in plasma and saliva were not as high as those given in previous reports [39, 40]. This was because HPV-16 was identified negative for all the enrolled HNSCC patients and one-third of the patients were in the early stages of the disease, which meant that less tumour DNA was released into the circulation and thus its levels were more difficult to determine [64].
The results of the present study indicate that sequencing of TAT, saliva and plasma for tumour DNA could potentially be incorporated into clinical examinations to complement current diagnostic modalities and inform clinical decision-making. Discrepancies in TAT evolutionary relationship with tumours indicate heterogeneity of possible mechanisms underlying HNSCC recurrence, awaiting further investigations.
Data availability
All data were collected locally and is accessible under reasonable requests.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Adelstein D, Gillison ML, Pfister DG, Spencer S, Adkins D, Brizel DM. et al. NCCN guidelines insights: head and neck cancers, version 2.2017. J Natl Compr Cancer Netw. 2017;15:761–70.
Mirghani H, Jung AC, Fakhry C. Primary, secondary and tertiary prevention of human papillomavirus-driven head and neck cancers. Eur J Cancer. 2017;78:105–15.
Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and-unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26:612–9.
Näsman A, Nordfors C, Holzhauser S, Vlastos A, Tertipis N, Hammar U, et al. Incidence of human papillomavirus positive tonsillar and base of tongue carcinoma: a stabilisation of an epidemic of viral induced carcinoma? Eur J Cancer. 2015;51:55–61.
Attner P, Du J, Näsman A, Hammarstedt L, Ramqvist T, Lindholm J, et al. The role of human papillomavirus in the increased incidence of base of tongue cancer. Int J Cancer. 2010;126:2879–84.
Chor JS, Vlantis AC, Chow TL, Fung SC, Ng FY, Lau CH, et al. The role of human papillomavirus in head and neck squamous cell carcinoma: a case control study on a southern Chinese population. J Med Virol. 2016;88:877–87.
Li H, Torabi SJ, Yarbrough WG, Mehra S, Osborn HA, Judson B. Association of human papillomavirus status at head and neck carcinoma subsites with overall survival. JAMA Otolaryngol Head Neck Surg. 2018;144:519–25.
Zengng J, Tang Y, Wu P, Fang X, Wang W, Fan Y, et al. Alcohol consumption, tobacco smoking, betel quid chewing and oral health associations with hypopharyngeal cancer among men in Central South China: a case–control study. Cancer Manag Res. 2019;11:6353–64.
Giraldi L, Leoncini E, Pastorino R, Wünsch-Filho V, de Carvalho M, Lopez R, et al. Alcohol and cigarette consumption predict mortality in patients with head and neck cancer: a pooled analysis within the International Head and Neck Cancer Epidemiology (INHANCE) Consortium. Ann Oncol. 2017;28:2843–51.
Poeta ML, Manola J, Goldenberg D, Forastiere A, Califano JA, Ridge JA, et al. The ligamp TP53 assay for detection of minimal residual disease in head and neck squamous cell carcinoma surgical margins. Clin Cancer Res. 2009;15:7658–65.
De-Vries N, Waal I, van der, Snow GB. Multiple primary tumors in oral cancer. Int J Oral Maxillofac Surg. 1986;15:85–87.
Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. Clinical implications of multicentric origin. Cancer. 1953;6:963–8.
Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–30.
Tabor MP, Brakenhoff RH, van Houten VM, Kummer JA, Snel MH, Snijders PJ, et al. Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001;7:1523–32.
Hittelman WN. Genetic instability in epithelial tissues at risk for cancer. Ann NY Acad Sci. 2001;952:1–12.
Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22.
Ryser MD, Lee WT, Ready NE, Leder KZ, Foo J. Quantifying the dynamics of field cancerization in tobacco-related head and neck cancer: a multiscale modeling approach. Cancer Res. 2016;76:7078–88.
Gabusi A, Morandi L, Asioli S, Foschini M. Oral field cancerization: history and future perspectives. Pathologica. 2017;109:60–65.
Boldrupldrup L, Gu X, Coates PJ, Norberg-Spaak L, Fahraeus R, Laurell G, et al. Gene expression changes in tumor free tongue tissue adjacent to tongue squamous cell carcinoma. Oncotarget. 2017;8:19389–402.
van Oijen MG, Slootweg PJ, Biomarkers P. Oral field cancerization: carcinogen-induced independent events or micrometastatic deposits? Cancer Epidemiol. 2000;9:249–56.
Bouguezzi A, Sioud S, Hentati H, Selmi J. Field cancerization in oral cavity: a review. J Clin Case Stud, Rev & Rep. 2020;2:1–5.
Kotb AAEW, Amer HW, Dahmoush HM. Detection of field cancerization in the clinically normal oral mucosa of cannabis and cigarette smokers. Indian J Public Health Res Dev. 2021;12:216.
Bansalnsal R, Nayak BB, Bhardwaj S, Vanajakshi CN, Das P, Somayaji NS, et al. Cancer stem cells and field cancerization of head and neck cancer-an update. J Fam Med Prim Care. 2020;9:3178–82.
Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun. 2017;8:1–14.
Jakubek YA, Chang K, Sivakumar S, Yu Y, Giordano MR, Fowler J, et al. Large-scale analysis of acquired chromosomal alterations in non-tumor samples from patients with cancer. Nat Biotechnol. 2020;38:90–96.
Meier JD, Oliver DA, Varvares MAN. Surgical margin determination in head and neck oncology: current clinical practice. The results of an International American Head and Neck Society Member Survey. Head Neck. 2005;27:952–8.
Singh S, Kumar M, Kumar S, Sen S, Upadhyay P, Bhattacharjee S, et al. The cancer-associated, gain-of-function TP53 variant P152Lp53 activates multiple signaling pathways implicated in tumorigenesis. J Biol Chem. 2019;294:14081–95.
Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61:112–23.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90.
Wang Y, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015;7:293ra104.
Vidal J, Muinelo L, Dalmases A, Jones F, Edelstein D, Iglesias M, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol. 2017;28:1325–32.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224.
Yang M, Topaloglu U, Petty WJ, Pagni M, Foley KL, Grant SC, et al. Circulating mutational portrait of cancer: manifestation of aggressive clonal events in both early and late stages. J Hematol. 2017;10:1–13.
Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133.
Shanmugam A, Hariharan AK, Hasina R, Nair JR, Katragadda S, Irusappan S et al. Ultrasensitive detection of tumor‐specific mutations in saliva of patients with oral cavity squamous cell carcinoma. Cancer. 2021;127:1576–89.
Ahn SM, Chan JY, Zhang Z, Wang H, Khan Z, Bishop JA, et al. Saliva and plasma quantitative polymerase chain reaction–based detection and surveillance of human papillomavirus–related head and neck cancer. JAMA Otolaryngol Head Neck Surg. 2014;140:846–54.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–95.
Lv J, Chen Y, Zhou G, Qi Z, Tan K, Wang H, et al. Liquid biopsy tracking during sequential chemo-radiotherapy identifies distinct prognostic phenotypes in nasopharyngeal carcinoma. Nat Commun. 2019;10:1–10.
Chen S, Zhou Y, Chen Y, Huang T, Liao W, Xu Y, et al. Gencore: an efficient tool to generate consensus reads for error suppressing and duplicate removing of NGS data. BMC Bioinform. 2019;20:1–8.
Frank MS, Fuß J, Steiert TA, Streleckiene G, Gehl J, Forster, M. Quantifying sequencing error and effective sequencing depth of liquid biopsy NGS with UMI error correction. Biotechniques 2021;70:226–32.
Li S, Lai H, Liu J, Liu Y, ** L, Li Y, et al. Circulating tumor DNA predicts the response and prognosis in patients with early breast cancer receiving neoadjuvant chemotherapy. JCO Precis Oncol. 2020;4:244–57.
Ai X, Lin Y, Liu A, **e C, Hu X, Zhao Q, et al. 52PD EGFR clonality and tumor mutation burden (TMB) analysis based on circulating tumor DNA (ctDNA) sequencing in advanced non-small cell lung cancer (NSCLC). J Thorac Oncol. 2018;13:S27–S28.
**ng P, Han X, Wang S, Liu Y, Yang S, Hao X. et al. Co‐mutational assessment of circulating tumour DNA (ctDNA) during osimertinib treatment for T790M mutant lung cancer. J Cell Mol Med. 2019;23:6812–21.
Zhang Y, Chang L, Yang Y, Fang W, Guan Y, Wu A, et al. Intratumor heterogeneity comparison among different subtypes of non-small-cell lung cancer through multi-region tissue and matched ctDNA sequencing. Mol Cancer. 2019;18:1–6.
Zhou J, Wang C, Lin G, **ao Y, Jia W, **ao G, et al. Serial circulating tumor DNA in predicting and monitoring the effect of neoadjuvant chemoradiotherapy in patients with rectal cancer: a prospective multicenter study. Clin Cancer Res. 2021;27:301–10.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Wu P, Wu H, Tang Y, Luo S, Fang X, **e C, et al. Whole-exome sequencing reveals novel mutations and epigenetic regulation in hypopharyngeal carcinoma. Oncotarget. 2017;8:85326–40.
Tada H, Takahashi H, Ida S, Nagata Y, Chikamatsu K. Epithelial–mesenchymal transition status of circulating tumor cells is associated with tumor relapse in head and neck squamous cell carcinoma. Anticancer Res. 2020;40:3559–64.
Wang Y, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015;7:293ra104.
Hamana K, Uzawa K, Ogawara K, Shiiba M, Bukawa H, Yokoe H, et al. Monitoring of circulating tumour-associated DNA as a prognostic tool for oral squamous cell carcinoma. Br J Cancer. 2005;92:2181–4.
Payne K, Spruce R, Beggs A, Sharma N, Kong A, Martin T, et al. Circulating tumor DNA as a biomarker and liquid biopsy in head and neck squamous cell carcinoma. Head Neck. 2018;40:1598–604.
Jaiswal G, Jaiswal S, Kumar R, Sharma A. Field cancerization: concept and clinical implications in head and neck squamous cell carcinoma. J Exp Ther Oncol. 2013;10:209–14.
Scholes AG, Woolgar JA, Boyle MA, Brown JS, Vaughan ED, Hart CA, et al. Synchronous oral carcinomas: independent or common clonal origin? Cancer Res. 1998;58:2003–6.
van Oijen MG, Leppers Vd Straat FG, Tilanus MG, Slootweg PJ. The origins of multiple squamous cell carcinomas in the aerodigestive tract. Cancers. 2000;88:884–93.
Partridge M, Emilion G, Pateromichelakis S, Phillips E, Langdon J. Field cancerisation of the oral cavity: comparison of the spectrum of molecular alterations in cases presenting with both dysplastic and malignant lesions. Oral Oncol. 1997;33:332–7.
Worsham MJ, Wolman SR, Carey TE, Zarbo RJ, Benninger MS, Van Dyke, DL. Common clonal origin of synchronous primary head and neck squamous cell carcinomas: analysis by tumor karyotypes and fluorescence in situ hybridization. Hum Pathol. 1995;26:251–61.
Califano J, Leong PL, Koch WM, Eisenberger CF, Sidransky D, Westra, WH. Second esophageal tumors in patients with head and neck squamous cell carcinoma: an assessment of clonal relationships. Clin Cancer Res. 1999;5:1862–7.
Lin L-H, Chang K-W, Kao S-Y, Cheng H-W, Liu, C-J. Increased plasma circulating cell-free DNA could be a potential marker for oral cancer. Int J Mol Sci. 2018;19:3303.
Yang W-Y, Feng L-F, Meng X, Chen R, Xu W-H, Hou J, et al. Liquid biopsy in head and neck squamous cell carcinoma: circulating tumor cells, circulating tumor DNA, and exosomes. Expert Rev Mol Diagn. 2020;20:1213–27.
Bellairs JA, Hasina R, Agrawal N. Tumor DNA: an emerging biomarker in head and neck cancer. Cancer Metastasis Rev. 2017;36:515–23.
Acknowledgements
Not applicable.
Funding
This work is supported by the National Natural Science Foundation of China (81302355, 31601159 and 81402336).
Author information
Authors and Affiliations
Contributions
Conception and design of work: P.W., Z.Y., Y.T., Y.F., S.Z., L.Y. and Y.H.; supervised the study: Y.T. and Y.H.; provided plasma, saliva and tissue specimens: C.X., J.Z. and X.L.; provided clinicopathological and survival data: C.X. and X.F.; pathological review of tumours: Y.L.; performed targeted-capture next-generation sequencing and provided somatic variant data: T.X.; performed most of the analysis of data: Z.Y. and N.K.; additional analysis: Z.Y., N.K. and T.X.; interpretation of the data: P.W. and Z.Y.; wrote the paper: P.W. and Z.Y.; reviewed and edited the paper: L.Y., X.X. and X.Y.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the ethical committee at **angya Hospital, Central South University. All methods were performed in accordance with the relevant guidelines, and informed consent was obtained. This study was performed in accordance with the Declaration of Helsinki.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, P., **e, C., Yang, L. et al. The genomic architectures of tumour-adjacent tissues, plasma and saliva reveal evolutionary underpinnings of relapse in head and neck squamous cell carcinoma. Br J Cancer 125, 854–864 (2021). https://doi.org/10.1038/s41416-021-01464-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01464-0
- Springer Nature Limited