
Abstract
Neddylation is the writing of monomers or polymers of neural precursor cells expressed developmentally down-regulated 8 (NEDD8) to substrate. For neddylation to occur, three enzymes are required: activators (E1), conjugators (E2), and ligators (E3). However, the central role is played by the ubiquitin-conjugating enzymes E2M (UBE2M) and E2F (UBE2F), which are part of the E2 enzyme family. Recent understanding of the structure and mechanism of these two proteins provides insight into their physiological effects on apoptosis, cell cycle arrest and genome stability. To treat cancer, it is therefore appealing to develop novel inhibitors against UBE2M or UBE2F interactions with either E1 or E3. In this evaluation, we summarized the existing understanding of E2 interaction with E1 and E3 and reviewed the prospective of using neddylation E2 as a pharmacological target for evolving new anti-cancer remedies.
Similar content being viewed by others

Introduction
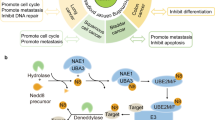
The neddylation pathway is a vital post-translational modification mechanism that plays a crucial role in several biological functions, including protein homeostasis, conformational change, and movement [1]. The process of neddylation is similar to that of ubiquitination. First, deneddylase 1 (DEN1) or ubiquitin C-terminal hydrolase domain 3 (UCH-L3) cleave the five amino-acid residues at the C-terminal end of the NEDD8 precursor, transforming it into mature NEDD8 (Fig. 1) [2, 3]. Once E1 recognizes the mature NEDD8, it initiates the neddylation process by utilizing the magnesium ion Mg2+ and adenosine triphosphate (ATP). To initiate conformational shifts in E1, which facilitate its binding to E2 and the subsequent transfer of NEDD8 to E2, two molecules of NEDD8 must initially bind to E1 [4]. After docking of E1’s adenylation domain with E2’s N-terminus, E1 releases the NEDD8 to E2 [5, 6]. In the final step, E2 works together with E3 ligases to transfer NEDD8 to the lysine residue of target molecules, which then activates downstream signaling pathways [7]. The NEDD8 modification is a reversible process carried out by deneddylase enzymes, including COP9 signalosomes (CSN), NEDD8-specific proteases (NEDP1), DEN1, and Ubiquitin carboxyl-terminal hydrolase 21, which remove NEDD8 from substrates [8,9,10,11]. While the classical neddylation pathway typically involves the addition of a single NEDD8 molecule to substrates, recent evidence suggests that polyneddylation can also occur and influence the function of substrates [12]. Several substrates of the neddylation pathway have been identified, including proteins in the CULLIN family, p53, the epidermal growth factor receptor (EGFR), E2F transcription factor 1 (E2F1), mouse double minute 2 (MDM2), and β-catenin [13, 14]. Currently, the neddylation pathway is known to be regulated by a single E1 enzyme or NEDD8 activating enzyme (NAE), two E2 enzymes (UBE2M/UBE2F), and over ten E3 enzymes [13].

Precursor NEDD8 with hydrolase is the first process before NEDD8 reacts with NAE. By actions of ATP and Mg2+, NAE is first loaded with one NEDD8 molecule. Following the same procedure, the second NEDD8 is attached with NAE. Double NEDD8 loaded NAE allows conjugating enzyme E2 binding and transferring NEDD8 from NAE’s active cysteine to E2’s cysteine. Next, by coordinating with E3 ligase, NEDD8 moves to the target protein. Finally, through the deneddylation process, NEDD8 is removed from the substrates.
The first E2 enzyme of the neddylation pathway to be discovered was the yeast-derived RUB1 conjugating enzyme (UBC12) [15]. Later, the human form of UBC12, known as UBE2M, was identified [5]. The second NEDD8 conjugating enzyme in humans, encoded by the UBE2F gene, was discovered in 2009 [16]. Initially thought to be mere “NEDD8 carriers” with limited functionality, modern research has revealed that E2 enzymes play essential roles in various physiological activities. UBE2M is overexpressed in several cancer types [17,18,19,20]. Normal physiological conditions rely on UBE2M for autophagy regulation, cell proliferation, stress granule assembly, and more [2D) [39]. Similarly, an acetylation at the N-terminus of UBE2F has been found to be responsible for its interaction with DCN3 [40]. Therefore, targeting the acetylated N-terminal end of UBE2F could lead to the development of novel and potent neddylation inhibitors similar to DI-591 (Fig. 2E).

A Classification of E2 conjugating enzyme based on N and C terminal extensions. B The crystal structure of UBE2M in green (PDB: 1Y8X) and UBE2F in the sky (PDB: 3FN1). C Virtually identified V30, F56, and C116 pockets of UBE2F for inhibitor binding. D Principle of develo** DI-591 and (E) Proposed idea of develo** E2 targeting inhibitor.
E2-based protein-protein interactions and their downstream effects on cancer
There is substantial evidence indicating that both UBE2M and UBE2F are crucial in controlling cancer cell growth, immune function, and survival. UBE2M is involved in the neddylation of various CULLIN and non-CULLIN proteins, making it a popular research subject. However, more investigation is needed on UBE2F. Recently, several reviews have provided detailed overviews of the roles of both UBE2M and UBE2F in various cancers [1, 41]. While both E2s receive NEDD8 from a single E1, they need different E3 ligases that can either support cell survival or lead to cell death. In this section, we will provide a summary of recent findings related to the functions of both E2s and their interactions with E1 and E3.
E2-E1 interaction
NAE is the only E1 enzyme involved in the neddylation process, which activates and transfers NEDD8 to either UBE2M or UBE2F. Analysis of the E2 structure revealed that the N-terminal extension of E2s is unique and necessary for docking in E1’s pocket [6]. However, further research is required to determine the situation when E1 selects between UBE2M or UBE2F. The E1 enzymes of neddylation consist of two heterodimeric subunits, amyloid precursor protein-binding protein 1, and UBL modifier activating enzyme 3 (UBA3) [42]. Proper interaction between the E2 enzymes and the UBA3 domain is crucial for the transfer of NEDD8 to E2 (Fig. 3A) [6, 36]. The N-terminal amino acids L4-F5-S6-L7 in UBE2M and M1-L2-T3-L4 in UBE2F bind to the docking groove of UBA3 [16], which follows an “HPR-HPR-AR-HPR” pattern (“HPR” representing hydrophobic residues, and “AR” representing any amino acid residue). Notably, hydrophobic residues F5 and L7 play a vital role in the proper docking of UBE2M’s N-terminal end with the large hydrophobic cavity of UBA3 (Fig. 3B, C) [36]. This unique property is exclusive to the E1-E2 neddylation interaction. Selective NAE-UBE2M or NAE-UBE2F interaction study is necessary to inhibit specific downstream neddylation. Therefore, targeting specific pockets in E1 or E2 that are involved in their interaction could lead to the development of new potential anti-cancer drugs.

A Interaction of E2 with E1. Appropriate interaction is necessary for NEDD8 transfer to E2. N-extension= N-terminal 26 residues of E2 (UBE2M). Improper docking of the E2 core domain or N-terminal extension prevented NEDD8 transfer from E1 to E2. B, C UBE2M’s N-terminal (purple) interaction with the hydrophobic pocket of UBA3 (cyan) and interacted amino acids, respectively (PDB:1TT5).
E2–E3 interactions
There are over 10 E3 ligases that interact with UBE2M or UBE2F to attach NEDD8 to specific substrates (Table 1). Of these, eight belong to the Ring finger domain-containing E3 class and they are RING-box protein 1 and 2 (RBX1 and RBX2), ring finger protein 111 and 168 (RNF111and RNF168), RNA polymerase II transcription factor B subunit 3 (TFB3), MDM2, casitas B lineage lymphoma (c-Cbl), and inhibitor of apoptosis (IAPs). Three E3s belong to the homologous to the E 6-AP carboxyl terminus (HECT) domain-containing E3 class and they are DCN1, itchy E3 ubiquitin ligase (ITCH), and smad ubiquitination regulatory factor 1 (SMURF1). The presence of HECT or RING domains distinguishes these two classes, which are crucial for their E3 ubiquitin ligase activity [43]. Tripartite motif-containing protein 40 (TRIM40) and F-box only protein 11 have also been identified as NEDD8 E3 ligases, however, their interaction with E2 conjugating enzyme was not reported yet [44, 45]. This section discusses how the interaction between E2 and E3 affects the neddylation of various types of substrates.
3.2.1. Interactions with Ring type E3 ligases
3.2.1.1. UBE2M–RBX1 interactions
RBX1 is a crucial component of the CRL1 complex, which is involved in the timely degradation of various substrates via ubiquitin ligation. In addition to its E3 ligase function for ubiquitin, RBX1 also interacts with UBE2M to facilitate the neddylation of CULLIN1-3, CULLIN4A, and CULLIN4B. This results in the formation of CRL1, CRL2, CRL3, CRL4A, and CRL4B, which promotes substrate ubiquitination and degradation [46]. RBX1’s structure consists of an N-terminal domain that binds to a substrate cullin’s C-terminal domain (CTD) and a C-terminal RING domain that attracts the UBE2M-NEDD8 intermediate. Upon neddylation, the RING domain stabilizes the UBE2M-NEDD8 in an active, closed conformation that can be nucleophilically attacked on the thioester link. To facilitate the transfer of NEDD8 to the substrate acceptor lysine residue, RBX1 brings the UBE2M-NEDD8 intermediate close to the bound substrate [47]. A recent study discovered that the UBE2M-RBX1 partnership is essential for regulatory T (Treg) cell fitness, indicating that neddylation maintains control of immunosuppressive function [46, 48]. Besides, by controlling various CRLs, RBX1-mediated neddylation controls the degradation of several critical substrates including p21, p27, CDT1, hypoxia-inducible factor 1-alpha (HIF1α), and NRF2, which are closely associated with cancer progression or inhibition.
Cancer promoting functions (CRL1 and CRL4): CULLIN 1 is a crucial part of the SCF (SKP1-CUL1-F-box protein) E3 ubiquitin ligase complex, which catalyzes the ubiquitination of various proteins for proteasomal degradation. Many proteins are degraded as a result of its activity, including transcription factors (Myc, IκBα, early growth response-1), cell cycle regulators (p21, p27, WEE1, CYCLINS D, and E), as well as apoptosis regulators (MCL-1, BimEL) [49]. CRL4A and CRL4B promoted the degradation of DNA damage-responsive protein CDT1 and cell cycle inhibitor p21 [50,51,52,53]. Besides, CRL4A promoted the IκB degradation to promote NF-κB activation which limits the apoptosis [54]. As UBE2M and RBX1 interaction is crucial for the assembly and activation of CRL1 and CRL4A/4B, inhibiting their formation could be a promising anticancer therapy. By targeting the inhibition of UBE2M and RBX1 interaction, it may be possible to develop new neddylation inhibitors with potential anticancer properties.
Cancer inhibiting functions (CRL2 and CRL3): CULLIN2 and CULLIN3 are the critical components of CRL2 and CRL3, respectively. When neddylation is inhibited in hypoxic conditions (favorable for tumor growth), CRL2 is deactivated, leading to an accumulation of HIF1α and increased stimulation of the HIF1 transcription factor [32, 55]. Activated HIF1 then promoted cell survivability, propagation, and angiogenesis by triggering various target genes responsible for regulating these processes [32]. This activation of HIF1 promotes the survival, proliferation, and angiogenesis of cells by inducing the expression of target genes that regulate these processes. Similarly, inactivated CRL3 results in the accumulation of NRF2, which has antioxidant properties that promote cell survival [33]. These findings show that the degradation of substrates by CRL2 and CRL3 limits cancer progression. Therefore, inhibiting the neddylation of CULLIN2 or CULLIN3 may not be beneficial for cancer treatment. Further research is needed to determine the specific conditions that result in the formation and activation of different CRLs.
UBE2F-RBX2 interactions
RBX2 is another NEDD8 E3 ligase in the ring protein family that activates CRL5 when interacting with UBE2F [56]. CRL5 regulates the ubiquitination and degradation of proteins such as Src kinase, the proapoptotic protein NOXA, and the tumor suppressor DEPTOR [UBE2M with RNF111 and RNF 168 interactions
During DNA damage, NEDD8 accumulates at damage sites, requiring the E2 enzyme UBE2M and the E3 ubiquitin ligase RNF111 [62]. RNF111 interacts with UBE2M and enhances the polyneddylation of histone H4, which promotes the development of RNF168 foci and DNA repair. Knockdown of both UBE2M and RNF111 reduces RNF168 activity and hampers DNA repair. Additionally, NEDD8 can bind to histone 2 A (H2A) and prevent it from being ubiquitinated and degraded [63]. RNF168 promotes both H2A ubiquitination and neddylation while being itself a substrate for NEDD8. The neddylation of RNF168 is required for its E3 ubiquitin activity, and the knockdown of UBE2M decreases RNF168’s ligase activity. These findings indicate that UBE2M, RNF111, and RNF168 work together to repair DNA damage. Limiting their interactions may prevent DNA damage repair and induce apoptosis. MDM2 is a well-known oncoprotein that regulates the tumor suppressor protein p53 by functioning as a ubiquitin E3 ligase [64]. However, recent studies have shown that MDM2 can also act as a NEDD8 E3 ligase after being phosphorylated by c-Src. Once phosphorylated, MDM2 recruits UBE2M to inactivate p53 by neddylation, making the inhibition of UBE2M-MDM2 interactions a promising strategy for cancer treatment in wild-type p53 cells [65]. The inhibitors of apoptosis proteins (IAPs) are endogenous inhibitors for programmed cell death. They contain two conserved regions- the baculovirus IAP Repeats (BIRs) and the really interesting new gene (RING) domains - that regulate caspases. The BIRs control the binding of IAPs to caspases, while the RING domain functions as a ubiquitin E3 ligase, causing auto-ubiquitination of IAPs and caspases. Research has demonstrated that IAPs can inhibit apoptosis by neddylation and deactivate caspase proteins using UBE2M. This suggests that targeting the interaction between UBE2M and IAP could be a promising strategy for develo** anti-cancer drugs[66]. The E3 ligase c-Cbl promotes the neddylation and polyubiquitination of proto-oncogene c-Src, leading to its degradation by the proteasome. This inhibits cell migration and survival by deactivating the PI3K-AKT pathway [67]. Additionally, c-Cbl inhibits leukemia development by neddylating and stabilizing the type II receptor (TβRII), which decreases the levels of transforming growth factor β (TGF-β) [68]. Moreover, the UBE2M and c-Cbl axis have been shown to promote the neddylation of the EGFR, leading to its sorting and degradation, which prevents the overactivation of EGFR in cancer cells [69]. These findings demonstrate that neddylation, which is regulated by the UBE2M-c-Cbl interaction, has a tumor-inhibitory effect. TFB3 is a component of TFIIH, a well-studied complex involved in nucleotide excision repair (NER) and transcriptional start. It was discovered that the yeast E2 conjugating enzyme UBC12 allowed TFB3 to neddylate yeast Rtt101 and cullin3 in vivo. The impact of this relationship on humans hasn’t yet been studied, though [70]. DCN1, a scaffold-type E3 ligase, plays a role in regulating the neddylation pathway and carcinogenesis and is often disrupted in several squamous cell carcinomas (SCCs) [6]. By binding to the acetylated N terminus of UBC12 and the winged-helix B domain of CULLIN1, DCN1 facilitates the neddylation of CULLIN1. Additionally, the DCN1-UBC12 interaction can be disrupted to selectively modulate CULLIN3 [43]. Overexpressing DCN1 promotes cancer cell proliferation and metastasis, as shown by its high mRNA and protein expression levels in various cancers [71]. Blocking DCN1 and E2 enzymes could inhibit selective neddylation, such as CULLIN3 neddylation. The co-crystal structure of UBC12-DCN1 reveals that DCN1 has a specific binding cavity that accommodates UBC12’s N-terminus [39]. Several inhibitors have targeted this interaction, which leads to the accumulation of CRL3 substrate NRF2 [47]. The compounds in this series may have broader use in treating diseases linked to the overproduction of reactive oxygen species. The discovery of the ITCH, E3 ubiquitin ligase, during genetic research on mouse coat color variations revealed that ITCH has protein-interacting WW domains for substrate recruitment and a HECT domain for ubiquitin transfer [72]. ITCH regulates a broad range of immunological responses, including T-cell activation and tolerance and T-helper cell development. Haiwen li et al. discovered that ITCH modifies JUNB by NEDD8 [73]. JUNB is a proto-oncogene that belongs to the AP-1 transcription factor family. To neddylate JUNB, ITCH binds to UBE2M and facilitates NEDD8 binding to the N-terminal site of JUNB. Neddylation attenuates the transcriptional activity of JUNB by increasing polyubiquitination and degradation [73]. In metastatic prostate cancer, the loss of JUNB resulted in increased cancer cell proliferation [74]. While research suggests that JUNB has both cell division-promoting and -inhibiting actions, its manifestation varies depending on the cell-cycle state and surrounding circumstances. Accordingly, the inhibition of JUNB neddylation and protection from degradation by blocking the UBE2M-ITCH interaction could be beneficial for certain cancer treatments. Nonetheless, further studies on various types of cancer are required to confirm this function. SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1) is another HECT-type E3 ligase that conducts auto-neddylation on several lysine residues by physically interacting with NEDD8 and UBE2M [75]. Neddylation increases the ubiquitin ligase activity of SMURF1, which promotes cancer cell proliferation, invasion, and increases tumor volume in a mice xenograft model. Meanwhile, SMURF1 autoubiquitination decreases when UBE2M is depleted. Elevated expression of NEDD8, SMURF1, NAE1, and UBE2M have been found to correlate with cancer development and poor prognosis [75, 76]. These findings suggest that selective inhibition of UBE2M-SMURF1 could potentially reduce the oncogenic activity of SMURF1.UBE2M-MDM2 interaction
UBE2M-IAP interaction
UBE2M- c-Cbl interaction
UBE2M-TFB3 interaction
Interactions of E2 with HECT type E3 ligases
UBE2M-DCN1 interactions
UBE2M-ITCH interaction
UBE2M-SMURF1 interaction
Inhibitors reported against E2-based protein-protein interaction
Currently, there are ongoing efforts to develop small molecules that target E2-based protein-protein interactions to inhibit neddylation. Several research groups have reported different types of inhibitors, as summarized in (Fig. 4 and Table 2). These include inhibitors targeting E2-E1 interactions and those targeting E2–E3 interactions, as reported in various studies.

The picture represents the inhibitors reported regarding E2 interaction inhibition with E1 or E3, “*” indicate the IC50 measurement according to western blot band observation.
Targeting E2-E1 interaction site
Disrupting the docking of E2 in the E1 enzyme can inhibit selective E1-E2 neddylation interaction. E2 family proteins are characterized by numerous protein-protein interactions site, rigid conformations, and the absence of druggable pockets [77]. The small size and dynamic nature of the catalytic site of E2 enzymes make it challenging for small molecules to interact with them. However, recent studies have identified new inhibitors of UBE2M and UBE2F. Yanan Li et al. identified two pockets in UBE2F (F56 and V30) responsible for UBE2F-NAE interactions that are suitable for virtual drug screening (Fig. 2C). They demonstrated that HA-1141, a small molecule, can inhibit the collaboration between F56 and NAE [38]. Later, Tiantian Xu et al. described HA-9104, another small molecule capable of inhibiting the interaction between UBE2F and NAE when targeted to the V30 pocket [78]. In addition, Yi-fan Chen et al. conducted cell-based natural product inhibitor screening and observed UBE2M neddylation changes upon inhibitor application. They identified Arctigenin as a selective UBE2M inhibitor that inhibited UBE2M and CULLIN neddylation with an IC50 value of less than 2.5 μM [16]. We discussed that UBE2M and UBE2F are accountable for the neddylation of CULLIN and other proteins in different ways. Despite UBE2M’s ability to neddylate most of the proteins, UBE2F neddylates only CULLIN5 [16]. Inhibiting both proteins genetically led to successful cancer treatment. Creating an artificial scarcity of UBE2M leaves tumor cells more vulnerable to chemotherapy, radiotherapy, DNA damage, and apoptosis. Similarly, UBE2F inactivation led to an increase in NOXA-dependent apoptosis and inhibited tumor growth. The E2s of neddylation also induce the immunosuppressive action of the tumor microenvironment, which disrupts cytotoxic T cell-mediated tumor eradication. In our discussion of E2, we have seen that E2 selectively binds with one E1 while interacting with more than ten E3. The inhibitors HA-1141, HA-9104, and arctigenin have already been found to inhibit E2-E1 interactions of the neddylation pathway. There is however a need for more intensive research to enhance the effectiveness of those inhibitors as anticancer drugs. We discussed that different E2-E3 interactions have different effects on cancer cells, depending on whether they are supportive or not. Hence, targeting selective E2-E3 interaction may lead to the discovery of selective anticancer drugs. As far as selective E2-based neddylation inhibitor discovery is concerned, the method mentioned here may prove useful.
In summary, the ongoing clinical studies for the NAE inhibitor MLN4924 demonstrate the promise of neddylation inhibition as an anticancer treatment. A develo** issue is that MLN4924’s global neddylation inhibition can be optimized by targeting specific downstream neddylation. We have talked about how E2–E3 interactions are crucial for the selective neddylation of substrates. While some substrate neddylation inhibits cancer progression, other substrate neddylation supports cancer proliferation. In this situation, focusing on certain E2-E3 interactions may result in the development of novel anticancer drugs with high therapeutic potential.
References
Zheng YC, Guo YJ, Wang B, Wang C, Mamun MAA, Gao Y, et al. Targeting neddylation E2s: a novel therapeutic strategy in cancer. J Hematol Oncol. 2021;14:57.
Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–91.
Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–92.
Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature. 2007;445:394–8.
Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274:12036–42.
Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8’s E1. Mol Cell. 2005;17:341–50.
Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell. 2011;19:168–76.
Chan Y, Yoon J, Wu JT, Kim HJ, Pan KT, Yim J, et al. DEN1 deneddylates non-cullin proteins in vivo. J Cell Sci. 2008;121:3218–23.
Gong L, Kamitani T, Millas S, Yeh ET. Identification of a novel isopeptidase with dual specificity for ubiquitin- and NEDD8-conjugated proteins. J Biol Chem. 2000;275:14212–6.
Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, et al. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–5.
Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, et al. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–43.
Ghosh DK, Ranjan A. HYPK coordinates degradation of polyneddylated proteins by autophagy. Autophagy. 2022;18:1763–84.
Zhou L, Jiang Y, Luo Q, Li L, Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer. 2019;18:77.
Wang B, Wang T, Zhu H, Yan R, Li X, Zhang C, et al. Neddylation is essential for β-catenin degradation in Wnt signaling pathway. Cell Rep. 2022;22:110538.
Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. Embo J. 1998;17:2208–14.
Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33:483–95.
Gao Q, Yu GY, Shi JY, Li LH, Zhang WJ, Wang ZC, et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget. 2014;5:7820–32.
Li L, Wang M, Yu G, Chen P, Li H, Wei D, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106:dju083.
Cukras S, Morffy N, Ohn T, Kee Y. Inactivating UBE2M impacts the DNA damage response and genome integrity involving multiple cullin ligases. PLoS One. 2014;9:e101844.
Zhang Y, Shi CC, Zhang HP, Li GQ, Li SS. MLN4924 suppresses neddylation and induces cell cycle arrest, senescence, and apoptosis in human osteosarcoma. Oncotarget. 2016;7:45263–74.
Zhang G-C, Yu X-N, Sun J-L, **ong J, Yang Y-J, Jiang X-M, et al. UBE2M promotes cell proliferation via the β-catenin/cyclin D1 signaling in hepatocellular carcinoma. Aging. 2020;12:2373–92.
Jayabalan AK, Sanchez A, Park RY, Yoon SP, Kang GY, Baek JH, et al. NEDDylation promotes stress granule assembly. Nat Commun. 2016;7:12125.
Lu G, Yi J, Gubas A, Wang YT, Wu Y, Ren Y, et al. Suppression of autophagy during mitosis via CUL4-RING ubiquitin ligases-mediated WIPI2 polyubiquitination and proteasomal degradation. Autophagy. 2019;15:1917–34.
Zhao B, Gao C, Shi D, Mao J, Zhao J, Guo L, et al. Knockdown of Nedd8‑conjugating enzyme UBE2M suppresses the proliferation and induces the apoptosis of intrahepatic cholangiocarcinoma cells. Oncol Rep. 2019;42:2670–9.
Brown JS, Lukashchuk N, Sczaniecka-Clift M, Britton S, le Sage C, Calsou P, et al. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015;11:704–14.
Zhao X, Jiang L, Hu D, Tang Y, Zhao G, Du X, et al. NPRL2 reduces the niraparib sensitivity of castration-resistant prostate cancer via interacting with UBE2M and enhancing neddylation. Exp Cell Res. 2021;403:112614.
Xu J, Lv G, Xu B, Jiang B. Overexpression of UBE2M through Wnt/β-Catenin signaling is associated with poor prognosis and chemotherapy resistance in colorectal cancer. Transl Cancer Res. 2020;9:5614–25.
Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–16.
Hopf LVM, Baek K, Klügel M, von Gronau S, **ong Y, Schulman BA. Structure of CRL7(FBXW8) reveals coupling with CUL1-RBX1/ROC1 for multi-cullin-RING E3-catalyzed ubiquitin ligation. Nat Struct Mol Biol. 2022;29:854–62.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6.
Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS, et al. Treatment-emergent mutations in NAEβ confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21:388–401.
Pugh CW, Ratcliffe PJ. The von Hippel-Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. 2003;13:83–9.
Taguchi K, Yamamoto M. The KEAP1-NRF2 system as a molecular target of cancer treatment. Cancers. 2020;13:46.
Burroughs AM, Jaffee M, Iyer LM, Aravind L. Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J Struct Biol. 2008;162:205–18.
van Wijk SJ, Timmers HT. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 2010;24:981–93.
Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, et al. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–35.
Cook WJ, Jeffrey LC, Sullivan ML, Vierstra RD. Three-dimensional structure of a ubiquitin-conjugating enzyme (E2). J Biol Chem. 1992;267:15116–21.
Li Y, Wang C, Xu T, Pan P, Yu Q, Xu L, et al. Discovery of a small molecule inhibitor of cullin neddylation that triggers ER stress to induce autophagy. Acta Pharm Sin B. 2021;11:3567–84.
Zhou H, Lu J, Liu L, Bernard D, Yang CY, Fernandez-Salas E, et al. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat Commun. 2017;8:1150.
Monda JK, Scott DC, Miller DJ, Lydeard J, King D, Harper JW, et al. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure. 2013;21:42–53.
Zhou L, Lin X, Zhu J, Zhang L, Chen S, Yang H, et al. NEDD8-conjugating enzyme E2s: critical targets for cancer therapy. Cell Death Discov. 2023;9:23.
Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL Jr., et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003;12:1427–37.
Kurz T, Chou YC, Willems AR, Meyer-Schaller N, Hecht ML, Tyers M, et al. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell. 2008;29:23–35.
Noguchi K, Okumura F, Takahashi N, Kataoka A, Kamiyama T, Todo S, et al. TRIM40 promotes neddylation of IKKγ and is downregulated in gastrointestinal cancers. Carcinogenesis. 2011;32:995–1004.
Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem. 2007;282:1797–804.
Wu D, Li H, Liu M, Qin J, Sun Y. The Ube2m-Rbx1 neddylation-Cullin-RING-Ligase proteins are essential for the maintenance of regulatory T cell fitness. Nat Commun. 2022;13:3021.
Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. 2020;10:746–65.
Zhou L, Zhang L, Chen S, Sun D, Qu J. Elevated Neddylation pathway promotes Th2 cells infiltration by transactivating STAT5A in hepatocellular carcinoma. Front Oncol. 2021;11:709170.
Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19:3215–25.
Jackson S, **ong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562–70.
Zhou L, Zhu J, Chen W, Jiang Y, Hu T, Wang Y, et al. Induction of NEDD8-conjugating enzyme E2 UBE2F by platinum protects lung cancer cells from apoptosis and confers to platinum-insensitivity. Cell Death Dis. 2020;11:975.
Yin L, Zhang J, Sun Y. Early growth response-1 is a new substrate of the GSK3β-FBXW7 axis. Neoplasia. 2022;34:100839.
Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283:29045–52.
Song Q, Feng S, Peng W, Li A, Ma T, Yu B, et al. Cullin-RING ligases as promising targets for gastric carcinoma treatment. Pharmacol Res. 2021;170:105493.
Frost J, Rocha S, Ciulli A. Von Hippel-Lindau (VHL) small-molecule inhibitor binding increases stability and intracellular levels of VHL protein. J Biol Chem. 2021;297:100910.
Zhang S, Sun Y. Cullin RING Ligase 5 (CRL-5): neddylation activation and biological functions. Adv Exp Med Biol. 2020;1217:261–83.
Zhao Y, **ong X, Sun Y. Cullin-RING Ligase 5: functional characterization and its role in human cancers. Semin Cancer Biol. 2020;67:61–79.
Cui D, Dai X, Gong L, Chen X, Wang L, **ong X, et al. DEPTOR is a direct p53 target that suppresses cell growth and chemosensitivity. Cell Death Dis. 2020;11:976.
Tan M, Xu J, Siddiqui J, Feng F, Sun Y. Depletion of SAG/RBX2 E3 ubiquitin ligase suppresses prostate tumorigenesis via inactivation of the PI3K/AKT/mTOR axis. Mol Cancer. 2016;15:81.
Luo J, Zou H, Guo Y, Tong T, Ye L, Zhu C, et al. SRC kinase-mediated signaling pathways and targeted therapies in breast cancer. Breast Cancer Res. 2022;24:99.
Laszlo GS, Cooper JA. Restriction of Src activity by Cullin-5. Curr Biol. 2009;19:157–62.
Ma T, Chen Y, Zhang F, Yang CY, Wang S, Yu X. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol Cell. 2013;49:897–907.
Li T, Guan J, Huang Z, Hu X, Zheng X. RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J Cell Sci. 2014;127:2238–48.
Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012;586:1390–6.
Batuello CN, Hauck PM, Gendron JM, Lehman JA, Mayo LD. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc Natl Acad Sci USA. 2015;112:1749–54.
Broemer M, Tenev T, Rigbolt KT, Hempel S, Blagoev B, Silke J, et al. Systematic in vivo RNAi analysis identifies IAPs as NEDD8-E3 ligases. Mol Cell. 2010;40:810–22.
Lee GW, Park JB, Park SY, Seo J, Shin SH, Park JW, et al. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene. 2018;37:5552–68.
Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol Cell. 2013;49:499–510.
Oved S, Mosesson Y, Zwang Y, Santonico E, Shtiegman K, Marmor MD, et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem. 2006;281:21640–51.
Rabut G, Le Dez G, Verma R, Makhnevych T, Knebel A, Kurz T, et al. The TFIIH subunit Tfb3 regulates cullin neddylation. Mol Cell. 2011;43:488–95.
Li J, Yu T, Yan M, Zhang X, Liao L, Zhu M, et al. DCUN1D1 facilitates tumor metastasis by activating FAK signaling and up-regulates PD-L1 in non-small-cell lung cancer. Exp Cell Res. 2019;374:304–14.
Aki D, Zhang W, Liu YC. The E3 ligase Itch in immune regulation and beyond. Immunol Rev. 2015;266:6–26.
Li H, Zhu H, Liu Y, He F, **e P, Zhang L. Itch promotes the neddylation of JunB and regulates JunB-dependent transcription. Cell Signal. 2016;28:1186–95.
Thomsen MK, Bakiri L, Hasenfuss SC, Wu H, Morente M, Wagner EF. Loss of JUNB/AP-1 promotes invasive prostate cancer. Cell Death Differ. 2015;22:574–82.
**e P, Zhang M, He S, Lu K, Chen Y, **ng G, et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun. 2014;5:3733.
Du MG, Liu F, Chang Y, Tong S, Liu W, Chen YJ, et al. Neddylation modification of the U3 snoRNA-binding protein RRP9 by Smurf1 promotes tumorigenesis. J Biol Chem. 2021;297:101307.
Zlotkowski K, Hewitt WM, Sinniah RS, Tropea JE, Needle D, Lountos GT, et al. A small-molecule microarray approach for the identification of E2 enzyme inhibitors in ubiquitin-like conjugation pathways. SLAS Discov. 2017;22:760–6.
Xu T, Ma Q, Li Y, Yu Q, Pan P, Zheng Y, et al. A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct Target Ther. 2022;7:354.
Chen YF, Liu RZ, Ying WW, Yang YN, **ang SF, Shao XJ, et al. Arctigenin impairs UBC12 enzyme activity and cullin neddylation to attenuate cancer cells. Acta Pharmacol Sin. 2022;44:661–9.
Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–8.
Scott DC, Hammill JT, Min J, Rhee DY, Connelly M, Sviderskiy VO, et al. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat Chem Biol. 2017;13:850–7.
Kim HS, Hammill JT, Scott DC, Chen Y, Min J, Rector J, et al. Discovery of Novel Pyrazolo-pyridone DCN1 Inhibitors Controlling Cullin Neddylation. J Med Chem. 2019;62:8429–42.
Zhou H, Zhou W, Zhou B, Liu L, Chern TR, Chinnaswamy K, et al. High-Affinity Peptidomimetic Inhibitors of the DCN1-UBC12 Protein-Protein Interaction. J Med Chem. 2018;61:1934–50.
Zhou H, Lu J, Chinnaswamy K, Stuckey JA, Liu L, McEachern D, et al. Selective inhibition of cullin 3 neddylation through covalent targeting DCN1 protects mice from acetaminophen-induced liver toxicity. Nat Commun. 2021;12:2621.
Wang S, Zhao L, Shi XJ, Ding L, Yang L, Wang ZZ. et al. Development of highly potent, selective, and cellular active Triazolo[1,5-a]pyrimidine-based inhibitors targeting the DCN1-UBC12 protein-protein interaction. J Med Chem. 2019;62:2772–97.
Zhou W, Ma L, Ding L, Guo Q, He Z, Yang J, et al. Potent 5-Cyano-6-phenyl-pyrimidin-based derivatives targeting DCN1-UBE2M interaction. J Med Chem. 2019;62:5382–403.
He Z-X, An Q, Wei B, Zhou W-J, Wei B-F, Gong Y-P, et al. Discovery of potent and selective 2-(Benzylthio)pyrimidine-based DCN1-UBC12 Inhibitors for Anticardiac Fibrotic Effects. J Med Chem. 2022;65:163–90.
Zhou W, Xu C, Dong G, Qiao H, Yang J, Liu H, et al. Development of phenyltriazole thiol-based derivatives as highly potent inhibitors of DCN1-UBC12 interaction. Eur J Med Chem. 2021;217:113326.
Bulatov E, Zagidullin A, Valiullina A, Sayarova R, Rizvanov A. Small molecule modulators of RING-Type E3 ligases: MDM and Cullin families as targets. Front Pharmacol. 2018;9:450.
Cai C, Wang S, Xu Y, Zhang W, Tang K, Ouyang Q, et al. Transfer learning for drug discovery. J Med Chem. 2020;63:8683–94.
Athanasios A, Charalampos V, Vasileios T, Ashraf GM. Protein-Protein Interaction (PPI) network: recent advances in drug discovery. Curr Drug Metab. 2017;18:5–10.
Zhou L, Jia L. Targeting protein neddylation for cancer therapy. Adv Exp Med Biol. 2020;1217:297–315.
Zhou Q, Lin W, Wang C, Sun F, Ju S, Chen Q, et al. Neddylation inhibition induces glutamine uptake and metabolism by targeting CRL3(SPOP) E3 ligase in cancer cells. Nat Commun. 2022;13:3034.
Mittler F, Obeïd P, Haguet V, Allier C, Gerbaud S, Rulina AV, et al. Mechanical stress shapes the cancer cell response to neddylation inhibition. J Exp Clin Cancer Res. 2022;41:115.
Olaizola P, Lee-Law PY, Fernandez-Barrena MG, Alvarez L, Cadamuro M, Azkargorta M, et al. Targeting NAE1-mediated protein hyper-NEDDylation halts cholangiocarcinogenesis and impacts on tumor-stroma crosstalk in experimental models. J Hepatol. 2022;77:177–90.
Zhang S, You X, Xu T, Chen Q, Li H, Dou L, et al. PD-L1 induction via the MEK-JNK-AP1 axis by a neddylation inhibitor promotes cancer-associated immunosuppression. Cell Death Dis. 2022;13:844.
Sekeres MA, Fram RJ, Hua Z, Ades L. Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J Clin Oncol. 2018;36:TPS7077.
Ferdosi SR, Taylor B, Lee M, Tang N, Peng S, Bybee R, et al. PTEN loss drives resistance to the neddylation inhibitor MLN4924 in glioblastoma and can be overcome with TOP2A inhibitors. Neuro Oncol. 2022;24:1857–68.
Salaroglio IC, Belisario DC, Bironzo P, Ananthanarayanan P, Ricci L, Digiovanni S, et al. SKP2 drives the sensitivity to neddylation inhibitors and cisplatin in malignant pleural mesothelioma. J Exp Clin Cancer Res. 2022;41:75.
Ni S, Liu Q, Chen X, Ding L, Cai L, Mao F, et al. Pro-senescence neddylation inhibitor combined with a senescence activated β-galactosidase prodrug to selectively target cancer cells. Signal Transduct Target Ther. 2022;7:313.
Yoshimura C, Muraoka H, Ochiiwa H, Tsuji S, Hashimoto A, Kazuno H, et al. TAS4464, a highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther. 2019;18:1205–16.
Funding
This work was supported by the National Key Research Program (No. 2018YFE0195100, China); The National Natural Science Foundation of China (No. 82020108030 and No. U21A20416, China); Basic and Frontier Technology Research Project of Henan Province (No. 212102310313, China); Youth Supporting Program from Henan Province (No. 2021HYTP060); Basic Research of the Key Project of the High Education from the Education Department of Henan Province (No. 22ZX008, China); Youth Supporting Program from Zhengzhou University (No. JC202044046); Key Research Program of Henan Province (No. 222102310054, China); Key Research Program of Higher Education of Henan Province (No. 22A350010, China); The Postdoctoral Research Grant in Henan province (No. 202102024 and No. 1902002, China); The Scientific Research Foundation of Zhengzhou University (No. 32210807, China)
Author information
Authors and Affiliations
Contributions
MAA M and YL collected all the information and wrote the entire manuscript. Y-PG, Y-CZ, YG, J-GS, and L-FZ took part in authoritative discussions and gave revisions to the manuscript. H-ML and L-JZ revised and finalized the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mamun, M., Liu, Y., Geng, YP. et al. Discovery of neddylation E2s inhibitors with therapeutic activity. Oncogenesis 12, 45 (2023). https://doi.org/10.1038/s41389-023-00490-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-023-00490-2
- Springer Nature Limited