
Abstract
Tooth development is a complex process that involves precise and time-dependent orchestration of multiple genetic, molecular, and cellular interactions. Ameloblastin (AMBN, also named “amelin” or “sheathlin”) is the second most abundant enamel matrix protein known to have a key role in amelogenesis. Amelogenesis imperfecta (AI [MIM: 104500]) refers to a genetically and phenotypically heterogeneous group of conditions characterized by inherited developmental enamel defects. The hereditary dentin disorders comprise a variety of autosomal-dominant genetic symptoms characterized by abnormal dentin structure affecting either the primary or both the primary and secondary teeth. The vital role of Ambn in amelogenesis has been confirmed experimentally using mouse models. Only two cases have been reported of mutations of AMBN associated with non-syndromic human AI. However, no AMBN missense mutations have been reported to be associated with both human AI and dentin disorders. We recruited one kindred with autosomal-dominant amelogenesis imperfecta (ADAI) and dentinogenesis imperfecta/dysplasia characterized by generalized severe enamel and dentin defects. Whole exome sequencing of the proband identified a novel heterozygous C-T point mutation at nucleotide position 1069 of the AMBN gene, causing a Pro to Ser mutation at the conserved amino acid position 357 of the protein. Exfoliated third molar teeth from the affected family members were found to have enamel and dentin of lower mineral density than control teeth, with thinner and easily fractured enamel, short and thick roots, and pulp obliteration. This study demonstrates, for the first time, that an AMBN missense mutation causes non-syndromic human AI and dentin disorders.
Similar content being viewed by others


Introduction
Ameloblastin (AMBN: MIM *601259) expression was first detected in the enamel organ and is the second most abundant enamel matrix protein, after amelogenin, known to have key roles in enamel formation.1,2 The extracellular matrix has a critical role in tissue development and homoeostasis by mediating cell growth, migration, differentiation, apoptosis, and gene expression.3,4 In the enamel extracellular matrix, AMBN is believed to participate in ameloblast attachment to the underlying enamel matrix5 and in modulation of enamel crystal growth,6 and mutations in this gene are already known to cause amelogenesis imperfecta (AI: MIM104530).7,8 AI is the term given to a heterogeneous group of disorders characterized by failure of normal amelogenesis, with a reported prevalence of between 1/700 and 1/14 000.9,10 Genes associated with non-syndromic AI encode proteins involved in the formation and maintenance of the develo** enamel matrix, including amelogenin (AMELX), enamelin (ENAM), kallikrein-related peptidase 4 (KLK4), matrix metallopeptidase 20 (MMP20), Golgi-associated secretory pathway pseudokinase (FAM20A), chromosome 4 open reading frame 26 (C4ORF26), and amelotin (AMTN).8,11,12,13,14,15,16,17 Other genes associated with non-syndromic AI include solute carrier family 24 member 4 (SLC24A4) and G-protein-coupled receptor 68 (GPR68), which are involved in ion transport18,19; laminin subunit beta 3 (LAMB3), integrin subunit beta 6 (ITGB6), collagen type XVII alpha 1 chain (COL17A1), and laminin subunit alpha 3 (LAMA3), which are involved in extracellular matrix adhesion20,21,22,23,24,25; family with sequence similarity 83 member H (FAM83H) and WD repeat domain 72 (WDR72), which are associated with intracellular vesicles26,27; acid phosphatase 4 (ACP4), which is a hydrolytic enzyme28,29; dentin sialophosphoprotein (DSPP), which is associated with dentin development30; and distal-less homeobox 3 (DLX3), which is associated with craniofacial development.31 Enamel abnormality is characteristic of mouse models with a deletion of Ambn exons 5 and 6 (Ambn −5,6/−5,6)32,33, and Ambn overexpression.33 For many years, there were no known mutations of AMBN associated with AI phenotypes. Recently, hypoplastic AI was found to be associated with homozygous exon 6 deletion8 and another family was identified with a novel homozygous splice-site mutation (c.532–1G > C).34 To date, no point mutations of AMBN have been found to be responsible for this disease.
Dentinogenesis is a highly ordered process in which the organic predentin matrix is progressively mineralized by ectomesenchymally derived cells called odontoblasts.35 The odontoblasts differentiate at the bell stage of tooth development, forming a single layer of cells lining the pulp cavity, where they secrete the organic predentin matrix into the underlying space.36 AMBN expression is also detected in pulpal mesenchymal cells, including preodontoblasts and young odontoblasts.37 AMBN is known to be involved in the mesenchymal–ectodermal interaction that precedes dentin and enamel secretion, and the sequential expression pattern of AMBN also acts as a signalling molecule.38 Currently, hereditary dentin disorders are divided into dentinogenesis imperfecta (DGI) and dentin dysplasia (DD).39 Recent genetic studies have shown that mutations of collagen type I alpha 1 chain (COL1A1), collagen type I alpha 2 chain (COL1A2), DSPP, secreted modular calcium-binding protein 2 (SMOC2), vacuolar protein sorting 4 homologue B (VPS4B), and ssu-2 homologue (SSUH2) cause dentin disorders.39,40,41,42,43,44,45,46,47,48,Full size image
Three extracted disused mandibular third molar teeth from IV:8, IV:2, and IV:9 were available for phenotypic characterization after obtaining their informed consent (the donors were approximately 20–22 years old). High-resolution computed tomography scanning (Fig. 3a–c and Supplemental Videos 1-3) of control tooth 1 from IV:8 and teeth 2 and 3 from IV:2 and IV:9 showed that tooth 1 exhibited a normal enamel layer in terms of thickness and mineral density (enamel density = (2.91 ± 0.06) g·cm‒3 compared with a range of (2.57–3.10) g·cm‒3 previously reported for enamel)51,52,53,54,55 and a normal dentin mineral density, whereas teeth 2 and 3 exhibited reduced enamel and dentin mineral density (Fig. 3c–d). Enamel thickness also appeared to be reduced. The pulp chambers were narrower with pulp chamber calcification (Fig. 3b). The roots were shorter, with small or obliterated root canals (Fig. 3b).

Tooth ultrastructural analyses. a–d High-resolution X-ray CT analysis of exfoliated teeth from control individual IV:8 and affected individuals IV:2 and IV:9. a 3D reconstruction of the tooth CT data: individual IV:8 and individuals IV:2 and IV:9. b 3D reconstruction of pulp chambers. Teeth of IV:2 and IV:9 exhibit thistle-shaped pulp chambers. c Typical CT sections through the teeth are presented using false colour calibrated with respect to mineral density to generate mineral density maps. Scale bar is marked in g/cm3. d Mean enamel and dentin mineral density for each tooth is also shown graphically. The control tooth of IV:8 exhibits enamel and dentin apparently normal in structure and density. Affected teeth of IV:2 and IV:9 exhibit enamel and dentin significantly reduced in mineral density compared with the normal tooth (P < 0.001). *P < 0.05, **P < 0.01, or ***P < 0.001. Videos of 3D-rendered CT data showing surface detail and the internal structure of all teeth are available as Supplementary Material. e–j SEM of representative exfoliated teeth. e Tooth of individual IV:8 exhibits normal enamel architecture comprising prisms (rods) of individual enamel crystallites. f Enamel of patient IV:9 tooth is in the normal range in terms of the order but characterized by fewer enamel prisms (rods) and wider inter-rod distances, h with occasional areas exhibiting a disturbed structure (asterisk). h The width of the DEJ of patient IV:9 is increased and the shapes of transitions within the DEJ zone are straight instead of sigmoidal. i SEM of the control tooth of individual IV:8 shows normal dentin structure. j The distribution of dentin tubules was not even and the size of the dentin tubular diameter was not consistent with the tooth of patient IV:9. j Most peritubular dentin is thicker and dentin tubes are smaller or obliterated completely compared to the control tooth.
When examined with scanning electron microscopy (SEM), the normal control teeth exhibited the prismatic structure characteristic of normal human enamel and dentin (Fig. 3e, g, i). SEM revealed that the mutation had little effect on the prismatic structure, but the numbers of enamel prisms of affected teeth were obviously reduced (Fig. 3f); the enamel also displayed distinct regions where the enamel structure was particularly disturbed (Fig. 3h). The outline of the dentin–enamel junction (DEJ) was changed: the width of the DEJ was increased and the structure of sigmoidal transitions within the DEJ zone, which firmly combine enamel and dentin together, became a straight line in appearance (Fig. 3h). Examination of the ultrastructure of dentin of affected teeth showed that the distribution of dentin tubules was not even and that the dentin tubular diameter was not consistent (Fig. 3j). This verified that there was an increase in the thickness of major peritubular dentin in mutant teeth, which reduced the size of or even obliterated the dentin tubules compared to control teeth (Fig. 3j).
Genetic analysis
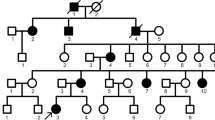
To identify the pathogenic variant responsible for the phenotype, we first screened for mutations in the coding and splice regions of the DSPP gene (exons 1–5). However, no mutations were identified. To confirm this result, long-range PCR was conducted with primers designed to amplify all exons, including the highly repetitive sequence in exon 5 of DSPP. Sequencing of the resulting product revealed that there were no mutations present within the DSPP exons or the flanking splice regions. In addition, we also analysed the copy number variations (CNVs) in the DSPP gene via array comparative genomic hybridization. Data analysis revealed no pathogenic CNVs in the DSPP gene in affected individuals in this family. Next, we performed exome sequencing with genomic DNA for affected individuals IV:2 and IV:9, and control IV:1 in collaboration with Genesky-Shanghai (China). Sequence analysis of all exons showed that 47 potentially pathogenic variants were shared by the two affected individuals and not by the unaffected individual. None of these variants were within genes known to be involved in AI, except for AMBN, or in DGI as identified by function, expression or animal model studies, including AMELX, ENAM, KLK4, MMP20, DSPP, SMOC2, VPS4B, and SSUH2. However, a novel heterozygous missense mutation (c.1069C > T) was identified in exon 13 of AMBN (NM: 016519, chr4:71472172), causing a proline to serine mutation at the conserved amino acid position 357 of the protein (Fig. 4a). The pathogenic variant of AMBN was validated by Sanger sequencing followed by co-segregation analysis in all affected and unaffected family members (Fig. 4b). The candidate region was confirmed by additional genotype data from microsatellite markers on 4q, and linkage analysis gave a maximum logarithm of odds score of 4.515 between markers D4S1541 and D4S1558 (Figs. 1 and 4a). Further, this nucleotide change was not detected in DNA samples from 185 normal unrelated controls and 15 dominant AI patients matched for Chinese ethnicity.

Interval map** and mutational analysis of AMBN. a Chromosomal location of the AMBN gene and the distribution of seven short tandem repeat (STR) markers. b Sanger sequencing result of the WT and the mutant. All the patients in this family carry a heterozygous mutation of AMBN (c.1069C > T). b This abnormal variation changes amino acid 357 of AMBN from a hydrophobic P to a hydrophilic S.
Functional analysis of mutant AMBN
The c.1069C > T mutation in AMBN changes the three-dimensional (3D) structure of the AMBN protein predicted by I-TASSER, leading to a loss-of-function protein estimated by Polyphen-2 (Fig. 5b). To evaluate the effect of the mutation, the cellular localization of AMBN was analysed using wild type (WT) and mutant constructs in human embryonic kidney (HEK) 293 cells. The result showed that the mutant AMBN was localized in the cytoplasm, which was the same as WT AMBN (Fig. 5c). However, the mutant protein exhibited aggregation in transfected HEK293 cells (Fig. 5c). Moreover, we made two WT and mutant constructs that were transiently expressed in human gingival fibroblasts (HGFs) and HEK293 cells. There was no obvious difference in mRNA expression between cells transfected with the mutated transgenic vector (ΔcAMBN - GFP) or normal transgenic vector (cAMBN - GFP) in either HGF (P > 0.05) or HEK293 cells (P > 0.05) (Fig. 5d). However, we found that expression of the aberrant protein was significantly increased compared with the WT AMBN protein in both HGF (P < 0.001) and HEK293 cells (P < 0.01) (Fig. 5e, f).

Effect of mutation on AMBN function. a Conservation analysis of this abnormal variation by Polyphen-2. The result showed that amino acid 357 of AMBN was highly conserved between different species. b The 3D structure of mutated AMBN was different from that of the wild-type predicted by I-TASSER. c Subcellular localization of AMBN in HEK293 cells. The mutant AMBN was localized in the cytoplasm similar to the wild-type protein, but the mutant protein exhibited aggregation in transfected HEK293 cells. d The mRNA expression level of AMBN in HEK293 and HGF cells. Mutant AMBN mRNA expression was no different than that of the wild type in either HEK293 cells (P > 0.05) or HGF cells (P > 0.05). e Western blot analysis of AMBN expression. The results showed that mutant AMBN was expressed at a higher level than the normal state. NC, negative control. f The protein expression level of AMBN in HEK293 and HGF cells. Compared with the wild type, the protein expression level of AMBN was higher in HEK293 cells (P < 0.01) and HGF cells (P < 0.001). Data in d and f are presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001
