

Abstract
Black phosphorus is an infrared layered material. Its bandgap complements other widely studied two-dimensional materials: zero-gap graphene and visible/near-infrared gap transition metal dichalcogenides. Although highly desirable, a comprehensive infrared characterization is still lacking. Here we report a systematic infrared study of mechanically exfoliated few-layer black phosphorus, with thickness ranging from 2 to 15 layers and photon energy spanning from 0.25 to 1.36 eV. Each few-layer black phosphorus exhibits a thickness-dependent unique infrared spectrum with a series of absorption resonances, which reveals the underlying electronic structure evolution and serves as its infrared fingerprints. Surprisingly, unexpected absorption features, which are associated with the forbidden optical transitions, have been observed. Furthermore, we unambiguously demonstrate that controllable uniaxial strain can be used as a convenient and effective approach to tune the electronic structure of few-layer black phosphorus. Our study paves the way for black phosphorus applications in infrared photonics and optoelectronics.
Similar content being viewed by others
Introduction
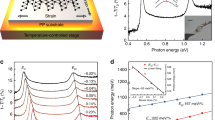
Since the isolation of graphene in 2004 (ref. 1), tremendous attention has been paid to the family of two-dimensional (2D) materials. Recently, black phosphorus (BP) was reintroduced as a new 2D material2,3,4,17,18,19. Previous photoluminescence (PL)14 and differential reflectance20 studies of BP are limited to the visible and near-infrared range, and are available only for thin BP layers with layer number <5. With the majority of the optical transitions expected in the mid- to near-infrared frequency range for few-layer BP, Fourier transform infrared spectrometer (FTIR)-based infrared spectroscopy is believed to be the superior characterization tool. However, up to date, such infrared study for mechanically exfoliated few-layer BP (<15L), with frequency ranging from the mid- to the near-infrared, is still lacking.
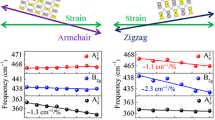
Here we systematically investigate the evolution of electronic structures in few-layer BP with layer number ranging from 2 up to 15, and report the experimental demonstration of highly tunable electronic structures in few-layer BP via controllable uniaxial strain21,22,23,24,25, using polarized infrared spectroscopy. For each few-layer BP, the infrared spectrum typically exhibits layer-dependent multiple optical resonances and can be readily served as its fingerprints. The infrared absorption shows strong polarization dependence, with strong optical resonances showing up in the AC direction. This dependence provides us a reliable way to determine the crystallographic orientation, which complements polarized Raman spectroscopy. For the latter, however, excitation wavelength and BP thickness complicate the polarization behaviour15,27, electrical field18, magnetic field28 and chemical do**. The rich band structures of few-layer BP and their potential large tunability promise a wide range of applications in infrared photonics and optoelectronics and  ) and one out-of-plane hop** parameter (t⊥) in a bilayer BP. (c) Extinction spectra (1−T/T0) of a 6L BP sample under varying tensile strains, with strain applied along the AC (red) and ZZ (blue) directions. The spectra are vertically offset. Here, 0.92%x (0.92%y) indicates applying 0.92% strain along the AC (ZZ) direction. The incident light is polarized along the AC direction. The dashed lines trace the shift in the transition energies. (d) The E11 and E22 peak energies as a function of tensile strains, the strain direction is along the AC (red) and ZZ (blue) directions, respectively. The solid lines are linear fits to the data.
) and one out-of-plane hop** parameter (t⊥) in a bilayer BP. (c) Extinction spectra (1−T/T0) of a 6L BP sample under varying tensile strains, with strain applied along the AC (red) and ZZ (blue) directions. The spectra are vertically offset. Here, 0.92%x (0.92%y) indicates applying 0.92% strain along the AC (ZZ) direction. The incident light is polarized along the AC direction. The dashed lines trace the shift in the transition energies. (d) The E11 and E22 peak energies as a function of tensile strains, the strain direction is along the AC (red) and ZZ (blue) directions, respectively. The solid lines are linear fits to the data.
As we know, BP is a highly anisotropic material, especially its mechanical properties exhibit strong crystallographic orientation dependence. For example, monolayer BP has been predicted to contract in the out of plane direction under tensile strain along the AC direction (positive Poisson’s ratio), whereas it expands under ZZ tensile strain (negative Poisson’s ratio)48. In addition, the Young’s modulus of monolayer BP in the ZZ direction is predicted to be 3.8 times larger than that in the AC direction10. With these in mind, let us now examine the direction dependence of the uniaxial strain effect on the electronic structures. The peak positions in Fig. 4c are summarized in Fig. 4d as a function of tensile strain in both AC and ZZ directions. Surprisingly, little difference is observed for the two distinct strain directions. For each strain, the peak positions of the sample stretched in both directions are almost always the same. From the linearly fitted lines, we can extract that the blueshift rate for E11 is 117 meV/% (AC direction) and 124 meV/% (ZZ direction), respectively, and 99 meV/% for E22 in both directions. It is not a coincidence for this specific sample. We have performed similar measurements on multiple samples, no strain direction dependence beyond experimental uncertainty has been observed, in sharp contrast to the giant anisotropic Raman response to uniaxial strain46,47.
Within the strain range from ɛ=0–0.92%, the optical bandgap (E11) can be continuously tuned from 0.54 to 0.65 eV for this 6L sample. Such large tunability is highly desirable for the application in high-sensitivity strain sensors. From the extracted peak shifting rates, we conclude that 1% tensile strain leads to a 23% increase of the bandgap for a 6L BP. The fractional change of the bandgap is even greater for thicker BP samples, given that the bandgap is smaller and the shift rate is similar for all layer thickness, as discussed later on. These observations are consistent with previous first-principle calculations for monolayer and bulk BP21,22,23,24.
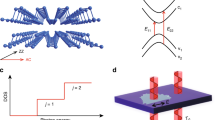
The observed electronic structure evolution can be understood within the tight-binding framework25,39,44,49. As strain is applied in the basal plane, it mostly affects the in-plane bonding. As a consequence, the strain effect (shift rates of the optical transitions) will be similar for mono- and few-layer BP, with little dependence on the layer thickness. Indeed, we observed a similar shift rate as that of the 6L BP sample for the E11 transition of a 3L BP sample, as shown in Supplementary Fig. 6. In fact, such phenomenon can be inferred from equation (1). In-plane strain has strong effect on Eg0 and smaller effect on γc−γv, which renders all optical transitions  similar strain dependence. Therefore, we only need to consider the strain effect on monolayer BP, whose band structure around the band-edge can be quite satisfactorily captured by two hop** parameters
similar strain dependence. Therefore, we only need to consider the strain effect on monolayer BP, whose band structure around the band-edge can be quite satisfactorily captured by two hop** parameters  and
and  (refs 39, 50), as schematically illustrated in Fig. 4b. The values of
(refs 39, 50), as schematically illustrated in Fig. 4b. The values of  and
and  have been obtained theoretically through density functional theory (DFT) calculations and tight-binding parameterization39. The bandgap of monolayer BP is expressed as
have been obtained theoretically through density functional theory (DFT) calculations and tight-binding parameterization39. The bandgap of monolayer BP is expressed as  , with
, with  and
and  . With this expression, we can immediately explain why it increases with tensile strain along the ZZ direction. When a small tensile strain is applied along the ZZ direction, the amplitude of the hop** parameter
. With this expression, we can immediately explain why it increases with tensile strain along the ZZ direction. When a small tensile strain is applied along the ZZ direction, the amplitude of the hop** parameter  will decrease, because the relevant bond length increases. At the same time,
will decrease, because the relevant bond length increases. At the same time,  has no change due to the fact that the relevant bond length remains unchanged, given that the bond orientation is perpendicular to the strain direction. Consequently, the bandgap Eg0 increases, given that
has no change due to the fact that the relevant bond length remains unchanged, given that the bond orientation is perpendicular to the strain direction. Consequently, the bandgap Eg0 increases, given that  is negative in the first place. A qualitative argument cannot be applied for strain along the AC direction, because both
is negative in the first place. A qualitative argument cannot be applied for strain along the AC direction, because both  and
and  decrease in this case. As demonstrated later, the overall amplitude of the bandgap still increases due to the dominant change of
decrease in this case. As demonstrated later, the overall amplitude of the bandgap still increases due to the dominant change of  over
over  in Eg0. The increase of the bandgap under uniaxial tensile strain for BP layers is in sharp contrast to mono- and few-layer MoS2, whose bandgap shrinks51.
in Eg0. The increase of the bandgap under uniaxial tensile strain for BP layers is in sharp contrast to mono- and few-layer MoS2, whose bandgap shrinks51.
More interestingly, the strain along the ZZ and AC directions have quantitatively the same effect on the optical transitions. This is very counter-intuitive, given the fact that BP is so anisotropic in terms of almost every property, in particular for the mechanical properties. To better understand this behaviour, we have to examine the strain effect more quantitatively within the tight-binding framework. With the hop** parameters obtained from DFT calculations39 (at zero strain,  ,
,  ) and, as a common practice, assume that they are inversely proportional to r2 (r is the bond length), one can directly derive the bandgap dependence on the strain for monolayer BP44,49:
) and, as a common practice, assume that they are inversely proportional to r2 (r is the bond length), one can directly derive the bandgap dependence on the strain for monolayer BP44,49:

with the unit of eV. In the equation, ɛx, ɛy and ɛz is the strain along x, y and z direction, respectively. It should be noted that for monolayer BP, ɛz reflects the change of the thickness of the single puckered sheet, that is, the vertical distance between atoms on the top and atoms at the bottom of the puckered sheet. In fact, in another 2D material—monolayer graphene, ɛz has no meaning since all atoms are on the same plane. According to equation (2), it is obvious that the pure ZZ strain (ɛy) is more effective to tune the bandgap than the pure AC strain (ɛx), which is consistent with our expectation. However, experimentally, due to the Poisson effect of the polyethylene terephthalate substrate, the strain is not truly uniaxial and deformations in other directions exist as well. BP layers stick to the underlying substrate and they deform in the same way as the substrate in the x–y plane. More specifically, the BP layer stretched in ZZ direction will slightly shrink in AC direction and vice versa. However, as the Poisson effect of the substrate has no orientation dependence, this effect cannot smear out the strain direction dependence shown in equation (2) and stretch along ZZ direction is still more efficient to change the electronic structure. On the other hand, in z direction, BP layers are free to shrink or expand and ɛz is non-zero. We attribute the observed lacking of orientation dependence for the strain effect to the change of the monolayer thickness under in-plane strain. More specifically, for a monolayer BP, the tensile strain along the AC direction leads to compression in z direction, which gives rise to additional bandgap enhancement (see equation (2), noticing that the coefficient of ɛz is negative). On the contrary, tensile strain along the ZZ direction results in expansion in the z direction due to a predicted negative Poisson’s ratio (νzy<0)48, which partially cancels out the ZZ strain effect. More quantitatively, through equation (2), we find out that a Poisson’s ratio difference (νzx−νzy) of ∼0.12 in z direction will smear out the difference of the effect for strain along the ZZ and AC directions. This is consistent with DFT calculations, which have obtained νzx and νzy, with their difference ranging from 0.07 to 0.3 (refs 24, 48). It should be noted that the different Poisson’s ratios in z direction for strain along the ZZ and AC directions are direct consequences of the hinge-like structure of a BP layer48.
Now we see that equation (2) semi-quantitatively describes the strain dependence of the electronic transitions in mono- and few-layer BP. However, the derived shift rate is smaller than what we observed experimentally. This discrepancy requires more effort in exactly determining the hop** parameters  and
and  , and reassessment of the assumption that t∝1/r2. If we keep
, and reassessment of the assumption that t∝1/r2. If we keep  and
and  values provided by Rudenko et al.39 and assume that t∝1/rβ, we find out that the index β∼6 can quantitatively account for the measured shift rates25.
values provided by Rudenko et al.39 and assume that t∝1/rβ, we find out that the index β∼6 can quantitatively account for the measured shift rates25.

