

Abstract
As we enter the era of precision medicine, characterization of cancer genomes will directly influence therapeutic decisions in the clinic. Here we describe a platform enabling functionalization of rare gene mutations through their high-throughput construction, molecular barcoding and delivery to cancer models for in vivo tumour driver screens. We apply these technologies to identify oncogenic drivers of pancreatic ductal adenocarcinoma (PDAC). This approach reveals oncogenic activity for rare gene aberrations in genes including NAD Kinase (NADK), which regulates NADP(H) homeostasis and cellular redox state. We further validate mutant NADK, whose expression provides gain-of-function enzymatic activity leading to a reduction in cellular reactive oxygen species and tumorigenesis, and show that depletion of wild-type NADK in PDAC cell lines attenuates cancer cell growth in vitro and in vivo. These data indicate that annotating rare aberrations can reveal important cancer signalling pathways representing additional therapeutic targets.
Similar content being viewed by others


Introduction
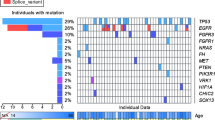
Large-scale efforts such as The Cancer Genome Atlas and International Cancer Genome Consortium (ICGC) are generating a compendium of genomic aberrations across major cancer types. Such studies are revealing the complexity of cancer genomes, which contain pathogenic driver aberrations and many more biologically neutral passenger events. While most cancers acquire one or more well-studied, high-frequency drivers, much less is known about which and how the abundant low-frequency variants contribute to tumour progression. Experimental assessment of low-frequency aberrations, in both under-characterized genes or among the long ‘tail’ mutations observed in known cancer genes, is difficult, given their large number and the fact that they may either directly drive tumour progression or indirectly influence tumour behaviour through modifying activities of other driver aberrations. Moreover, distinguishing driver events from passengers is further complicated by the fact that driver activity is shaped by the biological context of a given cancer, including its tissue type, microenvironment and other host determinants. Identifying rare driver aberrations therefore requires robust pipelines that allow context-specific functional prioritization of the thousands of potential targets emerging from Next Generation Sequencing (NGS) data.
Here we describe a gain-of-function (GOF) screening platform enabled by a High-Throughput Mutagenesis and Molecular Barcoding (HiTMMoB) method allowing cost-effective, rapid modelling and expression of molecularly barcoded mutant gene variants into open reading frame (ORF) expression clones for pooled functional screens. HiTMMoB permits annotation of mutation-activated oncogenes, whose identification is particularly desirable, given the efficacy of antibody and small molecule inhibitor therapies that may be tailored towards such proteins. Our approach complements gene depletion and knockout strategies by RNA interference (RNAi) and CRISPR/Cas9-based platforms, respectively, which have been tremendously successful for identifying and validating tumour suppressor genes and other genetic dependencies1,2. HiTMMoB builds on previous over-expression screen-based studies designed to identify wild-type ORFs whose expression promotes tumorigenesis3,4, drug resistance5 and other cancer cell phenotypes such as anchorage-independent growth6,7,8 among others. Such screens are made possible through efforts by the ORFeome collaboration (http://www.orfeomecollaboration.org/; refs 9, 10, 11) and others12 who have systematically cloned and sequenced-validated a large majority of expressed human transcripts.
In this study we use HiTMMoB to identify low-frequency mutational events in pancreatic ductal adenocarcinoma (PDAC) that serve as drivers or effectors for recurrent drivers such as KRAS, a proto-oncogene activated in >90% of PDAC cases13. We employed HiTMMoB to build PDAC aberration libraries, whose compositions were based on individual PDAC patient genomes and computational selection, to identify rare oncogenic events that, when expressed in a human pancreatic cell model and implanted into mice, promotes in vivo tumour formation. This approach revealed potent activity for aberrations in genes including NAD Kinase (NADK) that regulates NADP(H) homeostasis and cellular redox state. We show that mutant NADK provides GOF enzymatic activity, leading to a reduction in cellular reactive oxygen species (ROS) and tumorigenesis. In addition, we show that depletion of wild-type NADK in PDAC cell lines attenuated cancer cell growth in vitro and in vivo. We conclude that barcode (BC)-assisted, GOF screening has the potential to have an impact on the field of cancer target discovery as a whole, particularly with respect to identifying low-frequency somatic events by complementing structural characterizations of cancer genomes with functional annotation of gene aberrations identified from tumour sequencing programmes.
Results
HiTMMoB method
Successful translation of NGS data requires knowledge on which DNA aberrations represent actionable events, either for development or re-positioning of approved agents to target their activated pathways. To circumvent technical bottlenecks related to scaled construction of aberration expression clones, we developed a cost-effective HiTMMoB method (Fig. 1a) that employs: (1) two common PCR primers (P1/P2) flanking the ORF gene target and a third primer containing the desired mutation (PM), (2) a three-stage PCR reaction, whose design incorporates differential P1/P2 and PM melting temperatures54 using ɛ (solute)=2.0; ɛ (solvent)=78.0; probe radius=1.4 Å; [NaCl]=0.15 M and T=310 K. Amber 99SB partial atomic charges and Amber atomic radii were used for the electrostatic calculations. We used the Pymol graphics package for all illustrations.
NADK enzymatic assay
GST-NADK WT and GST-NADKI90F were cloned into pGEX-6 P-1 and expressed in One Shot BL21 (DE3; Life Technologies) for purification using Glutathione Agarose (Thermo Scientific) following the standard protocol. NADK and NADKI90F activity was measured spectrophotometrically with a coupled assay that reduces NADP generated by NADK into detectable NADPH using the enzyme glucose-6-phosphate dehydrogenase55. Data were analysed using GraphPad Prism 6 by means of Michaelis–Menten kinetics56.
NADK knockdown studies
NADK knockdown studies were performed using pLKO-shGFP (NT) or each of the following shRNAs available from the RNAi Consortium (TRC; Broad Institute): TRCN0000037702 (shNADK#4), TRCN0000199808 (shNADK#8), TRCN0000199040 (shNADK#10). shRNA target sequences are as follows: shGFP (NT): 5′-CAAGCTGACCCTGAAGTTCAT-3′; shNADK#4: 5′-GCATCAGCATCACTACCTCAT-3′; shNADK#8: 5′-GATGACATTTCCAATCAGATA-3′; shNADK#10: 5′-GAATAAGGAATTGAGTCCAGA-3′.
Additional information
Accession codes: The microarray data have been deposited in the NCBI GEO database under accession code GSE58055.
How to cite this article: Tsang, Y. H. et al. Functional annotation of rare gene aberration drivers of pancreatic cancer. Nat. Commun. 7:10500 doi: 10.1038/ncomms10500 (2016).
Accession codes
References
Moody, S. E., Boehm, J. S., Barbie, D. A. & Hahn, W. C. Functional genomics and cancer drug target discovery. Curr. Opin. Mol. Ther. 12, 284–293 (2010).
Mohr, S. E., Smith, J. A., Shamu, C. E., Neumuller, R. A. & Perrimon, N. RNAi screening comes of age: improved techniques and complementary approaches. Nat. Rev. Mol. Cell Biol. 15, 591–600 (2014).
Dunn, G. P. et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc. Natl Acad. Sci. USA 111, 1102–1107 (2014).
Wang, Y. et al. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. eLife 3, e01763 (2014).
Johannessen, C. M. et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138–142 (2013).
Shrestha, Y. et al. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene 31, 3397–3408 (2012).
Boehm, J. S. et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129, 1065–1079 (2007).
Pavlova, N. N. et al. A role for PVRL4-driven cell-cell interactions in tumorigenesis. eLife 2, e00358 (2013).
Rual, J. F. et al. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 14, 2128–2135 (2004).
Lamesch, P. et al. hORFeome v3.1: a resource of human open reading frames representing over 10 000 human genes. Genomics 89, 307–315 (2007).
Yang, X. et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661 (2011).
Temple, G. et al. The completion of the Mammalian Gene Collection (MGC). Genome Res. 19, 2324–2333 (2009).
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
Wu, W., Jia, Z., Liu, P., **e, Z. & Wei, Q. A novel PCR strategy for high-efficiency, automated site-directed mutagenesis. Nucleic Acids Res. 33, e110 (2005).
Furukawa, T. et al. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 148, 1763–1770 (1996).
Ouyang, H. et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am. J. Pathol. 157, 1623–1631 (2000).
Kuilman, T., Michaloglou, C., Mooi, W. J. & Peeper, D. S. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Singh, A. et al. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500 (2009).
Pollak, N., Dolle, C. & Ziegler, M. The power to reduce: pyridine nucleotides--small molecules with a multitude of functions. Biochem. J. 402, 205–218 (2007).
Sousa, C. M. & Kimmelman, A. C. The complex landscape of pancreatic cancer metabolism. Carcinogenesis 35, 1441–1450 (2014).
Wang, H. et al. Crystal structure of human NAD kinase. RCSB PDB (2011).
Rod, T. H., Radkiewicz, J. L. & Brooks, C. L. 3rd Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl Acad. Sci. USA 100, 6980–6985 (2003).
Meerbrey, K. L. et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl Acad. Sci. USA 108, 3665–3670 (2011).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
Kong, B., Qia, C., Erkan, M., Kleeff, J. & Michalski, C. W. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front. Physiol. 4, 246 (2013).
Hsieh, Y. C. et al. Enhanced degradation of dihydrofolate reductase through inhibition of NAD kinase by nicotinamide analogs. Mol. Pharmacol. 83, 339–353 (2013).
Fruehauf, J. P. & Meyskens, F. L. Jr Reactive oxygen species: a breath of life or death? Clin. Cancer Res. 13, 789–794 (2007).
Cui, X. Reactive oxygen species: the achilles' heel of cancer cells? Antioxid. Redox Signal. 16, 1212–1214 (2012).
Liou, G. Y. & Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 44, 479–496 (2010).
Pineda, C. T. et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728 (2015).
Liu, W., Cheng, S., Asa, S. L. & Ezzat, S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 68, 8104–8112 (2008).
Hirata, Y. et al. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 356, 244–248 (1994).
Kajimoto, Y. et al. Pancreatic islet cells express BST-1, a CD38-like surface molecule having ADP-ribosyl cyclase activity. Biochem. Biophys. Res. Commun. 219, 941–946 (1996).
Lee, H. C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 287, 31633–31640 (2012).
Lo Buono, N. et al. The CD157-integrin partnership controls transendothelial migration and adhesion of human monocytes. J. Biol. Chem. 286, 18681–18691 (2011).
Ortolan, E. et al. Functional role and prognostic significance of CD157 in ovarian carcinoma. J. Natl Cancer Inst. 102, 1160–1177 (2010).
Hingorani, S. R. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003).
Creighton, C. J., Nagaraja, A. K., Hanash, S. M., Matzuk, M. M. & Gunaratne, P. H. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA 14, 2290–2296 (2008).
Kammann, M., Laufs, J., Schell, J. & Gronenborn, B. Rapid insertional mutagenesis of DNA by polymerase chain reaction (PCR). Nucleic Acids Res. 17, 5404 (1989).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Pettersen, E. F. et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Dunbrack, R. L. Jr Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 12, 431–440 (2002).
Liu, J. et al. Crystal structures of an NAD kinase from Archaeoglobus fulgidus in complex with ATP, NAD, or NADP. J. Mol. Biol. 354, 289–303 (2005).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (1997).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001).
Lerner, F., Niere, M., Ludwig, A. & Ziegler, M. Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun. 288, 69–74 (2001).
Michaelis, L. & Menten, M. M. The kinetics of invertin action. 1913. FEBS Lett. 587, 2712–2720 (2013).
Acknowledgements
We thank Kristen Karlin and Trey Westbrook for providing and consulting on use of the pINDUCER vector system23 used to express KRASG12D. This project was supported in part by the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from the NIH/NCI grant (P30CA125123). This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT; RP120046), Lustgarten Foundation (RFP-B-042) and the 2014 Pancreatic Cancer Action Network—AACR Career Development Award (14-20-25-SCOT) by funding to K.L.S. This work was also supported by the NIH (UO1CA168394) by funding to K.L.S and G.B.M. T.D. was supported by a training grant from The Cullen Foundation. H.L. was supported by a training grant from CPRIT (RP140102). J.B.D. was supported by the American Cancer Society Joe and Jessie Crump Postdoctoral Fellowship. M.L. was supported by the NIH (R01CA138701).
Author information
Authors and Affiliations
Contributions
Y.H.T. and T.D. designed and performed experiments, produced figures and wrote the manuscript. P.T. designed kinase experiments and produced figures. J.R.B. supported kinase experiments. H.L., J.-W.O., N.N., R.M., J.B.D. and A.D. performed experiments. M.-C.G. provided patient tumour sequencing data. M.L., Q.C., H.Y. and M.I. performed pathology analysis. G.B.M. supported RPPA experiment. F.C. and C.J.C analysed microarray and RPPA data, and produced figures. J.K. performed computational modelling for NADK molecule and produced figures. M.E. performed the NGS run. K.E. and G.B.M. supported the NGS experiment. Z.C. and K.C. analysed the NGS data for BC enrichment analysis. K.L.S. supervised the study, designed experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8 and Supplementary Tables 1-2. (PDF 1606 kb)
Supplementary Data 1
Representative HiTMMoB data. (XLS 61 kb)
Supplementary Data 2
HPDE-iKRAS gene expression analysis. (XLSX 20952 kb)
Supplementary Data 3
Molecular Signatures Database Analysis. (XLSX 113 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tsang, Y., Dogruluk, T., Tedeschi, P. et al. Functional annotation of rare gene aberration drivers of pancreatic cancer. Nat Commun 7, 10500 (2016). https://doi.org/10.1038/ncomms10500
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10500
- Springer Nature Limited
This article is cited by
-
Restriction of extracellular lipids renders pancreatic cancer dependent on autophagy
Journal of Experimental & Clinical Cancer Research (2022)
-
The role of ROS in tumour development and progression
Nature Reviews Cancer (2022)
-
Precision oncology for breast cancer through clinical trials
Clinical & Experimental Metastasis (2022)
-
Comprehensive assessment of computational algorithms in predicting cancer driver mutations
Genome Biology (2020)
-
NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications
Signal Transduction and Targeted Therapy (2020)