
Abstract
To achieve the United Nations' Sustainable Development Goal (SDG7) of limiting global temperature rise to less than 1.5 °C, it is crucial to reduce non-renewable energy sources and curb the production of greenhouse gases like CO2. In this context, graphitic carbon nitride (g-C3N4) has emerged as a promising metal-free semiconductor photocatalyst for converting solar energy into clean fuels and valuable chemicals. However, there are challenges associated with g-C3N4, such as high electron–hole recombination, low photocurrent generation, limited specific surface area, and an absorption edge below 450 nm, which can be attributed to the arrangement of monomeric units. This review focuses on recent developments in designing single g-C3N4 as a metal-free catalyst through atomic-level do** and tuning surface chemical properties. Various do** techniques, including nonmetal and bi-nonmetal do**, as well as vacancy creation within the polymer framework and the effect of surface plasmonic nanoparticles, are explored as effective ways to fine-tune the polymer's conduction band (CB) edge potential, bandgap, and structural properties. The impact of do** and vacancy creation on the distribution of molecular orbitals, density of states (DOS), and adsorption energy on the polymer surface is investigated using computational calculations based on first principles and density functional theory (DFT). The review also examines the influence of do** on the photocatalytic reactions occurring in the polymer's CB, such as water splitting and carbon dioxide (CO2) reduction, and their selectivity in producing desired products. Last, the review summarizes the current challenges. It provides future perspectives on develo** metal-free photocatalysts, emphasizing the need to address unresolved structural, electronic, chemical, and optical properties to advance sustainable solutions. Overall, it is hoped that this review will inspire further research to unlock the full potential of metal-free photocatalysts and contribute to a more sustainable future.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Energy is a strategic commodity important for a country's growth, survival, and socio-economic development [1,2,3,4]. Our energy needs rely heavily on petroleum-based fuels, which have limited reserves and are not considered sustainable. Moreover, they have detrimental effects on the environment. Overcoming this challenge and ensuring a long-term energy supply while mitigating environmental impact necessitates exploring alternative energy sources [5,6,7]. Renewable energy sources like hydropower, wind, solar, and geothermal energy offer potential solutions. However, their widespread adoption faces obstacles due to geographical and technical constraints, which limit their acceptance in society [8,9,10]. To unlock the maximum energy potential from these resources and overcome these limitations, hydrogen (H2) is being investigated as a promising energy carrier. H2, with its efficient energy storage and delivery capabilities, is seen as a viable energy carrier for the future [11,12,13,14]. By utilizing H2 as an intermediary, we can effectively harness the energy generated from renewable sources and address the energy crisis. This approach opens up new possibilities for extracting the utmost energy from renewable resources, paving the way for a more sustainable and environmentally friendly energy landscape [15,16,17].
Harnessing solar energy provides a viable solution for meeting the demand for clean energy. Solar power is an abundant and renewable natural resource, with sunlight striking the Earth's surface at a rate approximately 130 million times greater than that generated by a single 500 MW power plant [18,19,20,21]. Utilizing solar energy through processes like photosynthesis [22], which converts sunlight into chemical energy, is essential for producing clean fuel. To fully use solar energy, we need to achieve specific vital objectives. The methods employed for solar energy conversion [23] and storage [25], biomass [26], and renewable sources like wind [27] and solar energy [28]. H2 fuel offers higher efficiency than gasoline [29] due to its lower volumetric energy density. However, the production of clean H2 still poses significant challenges that need to be addressed [30].
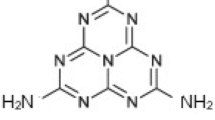
The groundbreaking research conducted by Fujishima and Honda [31] in 1972, which focused on using TiO2 as a semiconductor photoanode for splitting water through photocatalysis, provided hope for the generating, utilizing, and storing solar energy. However, TiO2's wide band gap of 3.2 eV and limited ability to absorb visible light hindered its efficiency in water splitting [32]. To overcome these challenges, scientists explored alternative photocatalysts with narrower band gaps, such as Fe2O3 [33], CdS [34], Bi2WO6 [35], BiVO4 [36], Ta2O5 [37], Ta3N5 [60,61,62], which involves introducing foreign elements or impurities, is a crucial technique for adjusting the physicochemical properties of g-C3N4. By do** one or two aspects into the polymer's framework, the band gap can be finely tuned [63], impacting its light absorption capacity and the values of its band edge potentials for various photocatalytic applications [64,65,66,67]. While there have been reports of metal do** [68,72] and their tendency to form covalent bonds by accepting electrons. Researchers have explored the do** of single nonmetals [73,74,75] and simultaneous do** of two nonmetals [76,77,78], which have shown significant enhancements in the activity of the bulk polymer. Given the recent progress in do** techniques, it is an ideal time to provide a comprehensive review of doped g-C3N4 systems for photocatalytic reduction reactions. While previous studies have covered aspects such as the design, development, and applications of g-C3N4, including the incorporation of both noble and non-noble metals [79,80,81], morphology orientation [82,83, In 1834, J. Liebig and J. J. Berzelius discovered [94] the formation of a pale-yellow, amorphous, and insoluble solid residue during the ignition of mercury (II) thiocyanate. They named this residue [95,96,97] "melon," now known as g-C3N4. Liebig determined its composition as (C2N3H) n through CHN analysis and identified melamine, melam, melem, and melon as the building blocks for the heptazine and triazine framework. Melon was synthesized by thermally polymerizing ammonium chloride and potassium thiocyanate. However, due to the insolubility and low reactivity of the s-heptazine derivatives, studying g-C3N4 faced challenges in the early years [98,99,100]. The focus was mainly on CHN elemental analysis (EA), and the structure remained unclear for a long time. In 1886, Klason proposed the formula C3N3(NH)3C3N3 without providing further details. Franklin, in 1922, reiterated the possibility of g-C3N4 as the final polymerization product and suggested potential structures, but without crystallography studies, his proposals were not widely accepted [101,102,103,104]. It was not until 1937 when Pauling and Sturdivant used X-ray crystallography to reveal the structure of the repeating unit, which consists of three fused 1,3,5-triazine rings known as tri-s-triazine [105]. In 1940, Redemann and Lucas confirmed Franklin's definition of the tris-s-triazine repeating unit by noting similarities between graphite and melon [106]. In 1989, Liu and Cohen [107] developed an empirical model and conducted ab initio calculations to predict the synthesis of new hard materials, including carbon–nitrogen compounds, with extreme hardness comparable to or greater than diamond. These theoretical predictions inspired [108,109,110] experimental researchers to explore synthetic methods. Further theoretical studies in the 1990s, particularly by Teter and Hamley, focused on the structure and stability of different g-C3N4 polymorphs (Fig. 2a). They predicted a cubic form of g-C3N4 with a higher bulk modulus than diamond. This theoretical work provided a foundation for experimental researchers to develop synthetic protocols. In 2001, Komatsu [111, 112] successfully synthesized Berzelius's melon and characterized it using various physicochemical techniques, such as X-ray diffraction, Raman spectroscopy, infrared spectroscopy, and laser desorption mass spectrometry. Kroke [113, 114] conducted a detailed structural characterization of a functionalized tri-s-triazine derivative and trichloro-tri-s-triazine the following year. They obtained bright yellow single crystals of trichloro-tri-s-triazine with a monoclinic space group (C2/c). DFT calculations supported a g-C3N4 structure based on tri-s-triazine, which was more stable than previously reported s-triazine structures by approximately 30 kJ mol−1. a Historical development and critical milestones of g-C3N4 from the 19th Century to the present day, based on established structure solutions: 1800s: Liebig, Berzelius, and Gmelin conducted early research on carbon and nitrogen compounds, laying the foundation for further exploration; 1930s: Pauling and Sturdivant made a significant breakthrough by identifying the heptazine unit (C6N7) as a crucial building block in carbon nitride compounds; 1990s: with the development of computational methods, scientists were able to simulate the structure of g-C3N4, providing valuable insights into the arrangement of carbon and nitrogen atoms; 2000: the structures of two crucial carbon nitride compounds, trichloroheptazine (C6N7Cl3) and melem (C6N7H6), were successfully determined, adding experimental evidence to support predicted structures. b Synthetic pathways for the polymer family of materials, with estimated reaction starting temperatures based on variable temperature XRD. Reproduced with permission from ref. 119. Copyright (2021) Wiley–VCH GmbH Jurgens conducted a significant study that involving synthesizing melem in large quantities, determining its crystal structure, and using spectroscopic techniques to validate the existence of a fundamental C-N–H molecule proposed earlier [115]. By subjecting cyanamide, ammonium dicyanamide, dicyandiamide, and melamine to thermal treatment at 450 °C for 5 h, a white-beige powder with a 60% yield was obtained. The molecule’s structure was confirmed through various methods, including single crystal X-ray diffraction (XRD), powder XRD, and 13C and 15N magic angle spinning nuclear magnetic resonance (MAS NMR) techniques. To gain further insights, the researchers performed calculations using B3LYP and MP2 level methods [116] implemented in Gaussian to determine the structural and vibrational properties of the molecule. The molecule's optimization in the extended state was carried out using Local Density Approximation (LDA) and Generalized Gradient Approximation (GGA) [117] implemented in VASP. The experimental and theoretical results agreed well regarding structural parameters and vibrational frequencies. Further investigations using temperature-dependent XRD revealed that the molecule transforms into a graphite-like C-N material when exposed to temperatures above 560 °C. The most stable phase of g-C3N4 has been successfully synthesized using nitrogen (N)-enriched organic compounds like cyanamide, dicyandiamide, melamine, and urea [118]. Wang et al.'s groundbreaking work in 2009 [129,130,131]. Using a covered crucible during fabrication played a crucial role in material formation. When the crucible was partially closed, a yellow solid powder was obtained. In contrast, an open crucible resulted in no material formation due to precursor decomposition caused by releasing gases during pyrolysis, which hindered the reaction. Therefore, the evolution of gases in a covered crucible acted as the driving force for polymerization. Moreover, the pyrolysis duration significantly affected the enhancement of active sites in the polymer. It was observed that the surface area of the polymer substantially increased when subjected to thermal polymerization for 0 to 240 min [132,171]. This process allows for precise adjustments in the semiconductor's characteristics. The concentration of dopants can vary, with low do** referring to a ratio of one dopant atom per 100 million intrinsic semiconductor atoms and high do** indicating a ratio of one dopant atom per ten thousand intrinsic semiconductor atoms [172,173,174]. The concept of do** in semiconductors has been known for a long time. As early as 1885, Bidwell discovered that impurities affected the properties of semiconductors [175]. Since then, extensive research has been conducted to develop do** techniques that enhance semiconductor performance for various applications [176,177,178,179]. In the field of nonmetal do** using g-C3N4 for photocatalysis, there are several advantages. First, do** increases the material's ability to absorb visible light, improving its performance [180, 181]. This occurs by enhancing the movement of excited electrons and holes while reducing their recombination rate. Do** achieves this by delocalizing π-conjugated electrons, causing the electron distribution in the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) to spread out. Second, do** allows precise adjustments to the polymer's CB edge potential. This makes the polymer suitable for enhancing selectivity in specific reactions, such as the reduction of CO2. The polymer's behavior can be tailored to facilitate desired reaction pathways by modifying the CB edge potential. Third, do** increases the material's active surface area by converting the bulk 3D polymer into 2D nanosheets. This generates more significant active sites and defects, providing more opportunities for catalytic reactions and improving overall efficiency. Finally, do** enhances the efficiency of photoelectrochemical processes by improving charge transfer dynamics at the solid/liquid interface. This facilitates the transfer of excited electrons to the CB, resulting in higher photocurrent than the undoped polymer. Boron (B) doped g-C3N4 was synthesized by heating a mixture of melamine and boron oxide. Boron oxide powders were dissolved in an ethanol (CH3CH2OH) solution, followed by the addition of melamine. The mixture was dried and then calcined at various temperatures. Analysis of the XRD patterns indicated that the insertion of B into the polymer's crystal structure caused a shift in the (002) peak towards higher angles. B dopants and N defects can be simultaneously introduced into g-C3N4 by a straightforward method. This involves calcining a mixture of g-C3N4 and sodium borohydride (NaBH4) in an N2 environment at various temperatures (Fig. 3a). By heating the g-C3N4 and NaBH4 mixture under controlled conditions, chemical reactions occur, and the dopants diffuse into the g-C3N4 structure. A N2 atmosphere prevents oxidation reactions by excluding oxygen (O2). XPS analysis of the B 1 s peak (Fig. 3b) confirmed the presence of B atoms incorporated into the g-C3N4 framework. X-ray absorption near-edge structure (XANES) measurements (Fig. 3c) showed distinctive 2p π* resonances related to specific N sites, namely triangular edge N (N1 site) and bridge N (N2 site). The N1/N2 peak intensity ratio indicated similar ratios of N atoms at N1 and N2 sites, suggesting that no N defects were introduced during B do**. At the same time, the B dopants were located at carbon (C) sites. DFT calculations were conducted to examine the electronic properties of g-C3N4 (Fig. 4a) and B doped g-C3N4 (Fig. 4d). The calculations revealed that the CB primarily consisted of C 2p and N 2p orbitals. In contrast, the valence band (VB) was mainly composed of N 2p orbitals (Fig. 4b and c). A midgap energy level involving C 2p, N 2p, and B 2p orbitals appeared around 1.5 eV above the VB (Fig. 4e and f). This midgap state contributed to increased absorption and was consistent with the observed absorption tail in the BHx samples. In terms of molecular orbitals, the HOMO and LUMO of pristine g-C3N4 (Fig. 4g) were uniformly distributed over the heptazine rings due to the material's symmetrical structure. However, in B doped g-C3N4 (Fig. 4h), the charge density was redistributed, resulting in the VB and CB’s downshifting and midgap states associated with dopants or defects. This redistribution of charge carriers facilitated spatial separation of charge localization sites, minimizing back reactions and enhancing the material's photocatalytic activity [182]. B do** increased the electrical conductivity of g-C3N4 and induced spin polarization. The do** concentration influenced the coupling configuration of the net spin and the resulting magnetism. However, the distribution of net spin at the active site remained unaffected by the do** concentration, indicating that the catalytic capacity was independent of the do** level [183]. Time-domain DFT provided a detailed mechanistic understanding of charge separation and recombination processes, which are crucial for assessing photocatalytic performance. Dopant states expanded the visible light absorption range and reduced nonradiative lifetimes compared to the pristine polymer. B do** at C exhibited a longer charge recombination time among the do** systems due to weaker electron–phonon coupling between the VB maximum and trap states. This finding suggests that B do** is the most suitable option for photocatalytic applications based on quantum dynamics processes [184]. a Schematic representation of the process for preparing B doped g-C3N4, b proposed structural changes in the heptazine units of g-C3N4 resulting from thermal treatment with NaBH4, c high-resolution spectra of B1s X-ray photoelectron spectroscopy showing g-C3N4 and B doped g-C3N4 (BHx, x = 300, 350, 400, 450, 500), d nitrogen K-edge X-ray absorption near-edge structure (XANES) spectra comparing g-C3N4, CN–B, CN–H, and BH400. Reproduced with permission from ref. 182. Copyright (2019) WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim a Diagram illustrating the structure model of pristine g-C3N4 and d B doped g-C3N4 with B2-site dopants, b calculated band structures shown for pristine g-C3N4, e calculated band structures displayed for B doped g-C3N4 with B2-site dopants, c density of states (DOS) plot corresponding to pristine g-C3N4, f density of states (DOS) plot corresponding to B doped g-C3N4, g visualization of the distribution of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in pristine g-C3N4, h visualization of the distribution of the HOMO and LUMO in B doped g-C3N4 with B2-site dopants, carbon atoms are depicted as grey spheres, nitrogen atoms as blue spheres, hydrogen atoms as white spheres, and boron atoms as red spheres. Reproduced with permission from ref. 182. Copyright (2019) WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim In addition to incorporating B atoms, C do** can also modify the electronic structure of the polymer, resulting in improved electron mobility and increased ability to absorb visible light. Researchers developed a hydrothermal calcination method to introduce C into the polymer by using melamine and sodium alginate as precursors (Fig. 5a). By adjusting the amount of sodium alginate, they investigated how the C content affects the structure and photocatalytic properties of the material. The presence of C do** was confirmed by observing higher intensity peaks in the C 1 s (at 284.1 eV) and N 1 s (at 399.2 eV) spectra of the doped polymer. The C doped material exhibited significantly stronger absorption in the visible light range (450 nm to 700 nm), which can be attributed to the intrinsic modulation of electron behavior and the band gap (Fig. 5b). Moreover, C do** reduced the rate of charge recombination, as indicated by fluorescence and time-resolved fluorescence spectroscopy. This suggests that C do** enhances the efficiency of proton reduction reactions, leading to more effective H2 production than the bulk polymer. These improvements can be attributed to the increased specific surface area, improved visible light absorption, and enhanced electron transport facilitated by the hollow tubular structure of the material [185]. Another approach involved mixing melamine with biomass-based kapok fiber to achieve C modification of g-C3N4 through a one-step pyrolysis process. C do** improved charge separation and exhibited superior photocatalytic activity under visible light [186]. Additionally, a C-rich g-C3N4 material with cross-pore channels was synthesized through the supramolecular self-assembly of melamine and 2-hydroxypropyl-β-cyclodextrin (Fig. 5c). This material demonstrated a larger surface area enhanced light-harvesting ability, faster separation of photoinduced carriers through abundant C rings, a narrower band gap, good photo response current (Fig. 5d) and excellent photocatalytic performance [188]. In 2012, scientists used hydrothermal method to synthesize oxygen (O) doped g-C3N4 by introducing hydrogen peroxide (H2O2) as a dopant precursor. The catalyst's N–C–O species was identified through an O 1 s core level detected at 531.6 eV. By incorporating O, which was generated through the pyrolysis of melamine precursors modified with H2SO4 and HNO3, the researchers observed an unusual improvement in light absorption and charge separation within the polymer. This enhancement was attributed to increase in the C–O–C content by replacing N atoms at N2c sites. The intensity of EPR signals gradually decreased, indicating a reduction in the concentration of unpaired electrons on C atoms. This decrease formed C–O–C bonds in aromatic rings, resulting in distinct morphologies. Additionally, the do** caused a shift in the potential of the CB edge to a more negative value, facilitating a more efficient photocatalytic reduction reaction [189]. Using a "calcination-hydrothermal-calcination" method involving melamine and oxalic acid (Fig. 6a), researchers successfully demonstrated the formation of a tube-in-tube structured nanoreactor composed of O doped g-C3N4 with a broom-like appearance. Analysis using transmission electron microscopy (TEM) revealed that the nanotube had a length of 18.5 μm and a wall thickness of 13.5 nm (Fig. 6b). The ultrathin wall of the nanotube played a crucial role in increasing light scattering within the hollow cavity, promoting the separation and migration of photo-generated charges. Furthermore, the confinement effect of this nanoreactor facilitated the rapid transfer of reactants, thereby accelerating the reaction rate. The specific morphology design of the tube-in-tube nanoreactor proved vital in enhancing the photocatalytic performance. Finite-difference time-domain (FDTD) simulations showed that the hollow structure allowed light to penetrate the interior, generating resonance. Under 420 nm wavelength irradiation, the hollow tube structure exhibited a strong local electric field induced both at the edge and inside, in contrast to the weak electric field observed only at the edge in the bulk polymer. DFT calculations provided insights into the effects of O do** and the formation energy associated with substituting O atoms at various N sites. The optimized band structure and DOS analysis revealed that the CB primarily formed from C 2p and N 2p orbitals, while the VB consisted mainly of N 2p orbitals. The calculations indicated a reduction in the band gap and a shift in the CB band edge upon O do**. The charge density around O atoms (Fig. 6c) increased in the O doped catalyst due to their strong interaction with neighboring atoms. Bader charge analysis at different sites confirmed a decrease in the charge density of C atoms near O atoms and an increase in the electron density of adjacent N atoms, suggesting a reorganization of charges within the doped catalyst compared to the undoped one (Fig. 6d). This redistribution of charges resulted in the creation of an internal electric field, facilitating the separation of electron–hole pairs [190]. Additional research using an ab initio approach has provided further insights into the positive effects of O do** in monolayer g-C3N4. Incorporating O dopants weakens the covalent nature of O-C bonds and introduces a complex ionic-covalent character in O-N bonds. This do** process enhances the material's ability to absorb visible light, creating an intrinsic semiconductor behavior and preventing recombination centers by distributing the HOMO and LUMO [191]. In another study, scientists successfully demonstrated the production of 2D amorphous O doped g-C3N4 by reacting the polymer with H2O2 and polyvinylpyrrolidone (PVP) in CH3CH2OH (Fig. 6e). Subsequently, the material was treated with supercritical CO2, leading to the formation of exfoliated nanosheets ranging in size from sub-micrometer to several micrometers, as observed through TEM and atomic force microscopy (AFM) [192]. Furthermore, a one-dimensional O doped g-C3N4 nanorod was synthesized using a hydrothermal method assisted by H2O2. By examining time-dependent FESEM and TEM images of the samples obtained at various hydrothermal times, it was observed that the nanosheets gradually dissolved over a continuous 24-h hydrothermal reaction, resulting in the formation of short and long nanorods and ultimately pure rod-like O doped g-C3N4 (Fig. 6f). The proposed mechanism suggests that this transformation occurs through the formation of smaller heptazine units with a higher number of -NH and -OH groups, which subsequently assemble through intermolecular H2 bonding and interlayer p-p stacking. O do** plays a crucial role in tuning the material's absorption band, enabling it to absorb light up to 537 nm and causing a color change from yellow to ivory white. Furthermore, the flat band potential shifts from -1.2 to -1.3 eV, significantly enhancing the efficiency of photocatalytic reactions. DFT calculations indicate that electron distribution around the Fermi level differs between the O doped system and the bulk polymer, where the Fermi level is situated within the middle of the band gap. O do** also prolongs the lifetime of excited electrons, as revealed by time-resolved photoluminescence (TRPL) spectra, and improves charge transfer efficiency, as demonstrated by photocurrent and impedance measurements [193]. To prepare O doped g-C3N4, a simple thermal-oxidative exfoliation method was employed. The process involved subjecting bulk g-C3N4 to two consecutive calcination steps at 600 °C for 1 h each, resulting in do**. X-ray diffraction analysis showed a decrease in the intensity of the (100) and (002) peaks with longer calcination times, and the (100) peak nearly disappeared after do**, leading to restacking along the Z direction. XPS and EA confirmed an increase in the atomic weight percentage of O from 3.54 to 4.73%, indicating the successful incorporation of O dopants. UVDRS revealed that all samples exhibited good absorption ability in the 200–300 nm wavelength range. Additionally, the bandgaps of the material increased from 2.5 to 2.9 eV. This increase in bandgap can be attributed to the quantum confinement effect resulting from the formation of thin nanosheets. The fluorescence intensity of the material also increased and exhibited a blue shift, further supporting the quantum size effect of the catalyst. The presence of O dopants led to greater photocurrent and lower impedance values, indicating reduced electron–hole recombination and improved charge transfer mobility. DFT studies supported these findings, showing that as the thickness of the polymer decreased, the bandgap became wider, consistent with the experimental results. Furthermore, O do** introduced impurity levels in the middle of the bandgap, facilitating the charge transition from the valence band maximum (VBM) to the do** level. This enhancement resulted in better performance for CO2 photoreduction [194]. a Oxalic acid and melamine formed connections through acid–base interactions, and during the calcination process, a portion of the mixture decomposed, releasing gases such as CO2 and NO2. Meanwhile, another portion underwent polycondensation to produce g-C3N4, incorporating oxygen atoms into the tri-s-triazine structure. Upon hydrothermal treatment under high temperature and pressure, water exhibited properties of a protonic acid, effectively breaking the hydrogen bonds between layers. As a result, the polymer tri-s-triazine structures underwent hydrolysis, yielding smaller molecular units. These smaller units were subsequently reassembled into a broom-shaped supramolecular precursor. b TEM images of O doped g-C3N4. Optimized structure and charge density difference of c g-C3N4 and d O doped g-C3N4. Reproduced with permission from ref. 190. Copyright (2023) Elsevier B.V. e At elevated temperatures, H2O2 molecules undergo decomposition, generating active radicals such as ·OH, HO2·, and H+. The tri-s-triazine-based compounds react with H+, and the protonation of g-C3N4 facilitates the grafting of hydroxyl radicals onto carbon, resulting in the hydroxylation modification. A high concentration of supercritical CO2 molecules induces a strong attacking interaction due to shear strain. Consequently, the C-N = C(-NH2+) bonds break, leading to the formation of nitrogen vacancies. Subsequently, O atoms replace nitrogen sites and establish covalent C–O–C bonds within the conjugated structure. Reproduced with permission from ref. 192. Copyright (2021) Zhengzhou University. f FESEM image of O doped g-C3N4 upon H2O2 treatment. Reproduced with permission from ref. 193. Copyright (2019) The Royal Society of Chemistry In 2010, researchers achieved a significant milestone by successfully incorporating phosphorus (P) atoms into the g-C3N4 framework through substitutional do**. They used a mixture of dicyandiamide and 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid as the source of P, with concentrations of 10 and 30 wt%. This mixture was then heated to create the doped catalyst. The researchers used various techniques to analyze the doped material, including XPS and 31P NMR. The XPS analysis showed that the presence of P-N coordination was indicated by the P2p peaks appearing at 133.5 eV, which is higher than the typical range for P–C coordination. Interestingly, no fluorine (F) peaks were observed in the XPS spectrum, indicating that the P source did not introduce F atoms to the material. The 31P NMR investigations provided additional insights, revealing distinct signals between 0 and -40 ppm, attributed to forming P-N bonds. Do** the material with lower amounts of P allowed most of its structural features to be retained. However, significant changes in the electronic properties were observed. The doped material changed color from yellow to brown and showed a substantial increase in electrical conductivity, reaching up to four orders of magnitude without light exposure. Additionally, the photocurrent values demonstrated a fivefold enhancement compared to the bulk material, indicating improved photocatalytic properties [195]. Novel porous P doped g-C3N4 nanosheets were fabricated by combining P do** and thermal exfoliation strategies by varying the mass ratio of 2-aminoethylphosphonic acid as a dopant precursor with melamine. The creation of macropores is due to the evolution of ammonia gas produced from the decomposition of the dopant as confirmed by the SEM and TEM. The thermal exfoliation gradually oxidized the heptazine units, reduced the thickness to 5–8 nm, and exposed these macropores with 90–300 nm sizes. Porous nanosheet architecture could enhance the surface area with higher active sites, facilitate the rapid transfer of photogenerated charge carriers and easy diffusion of reactant and product molecules by enhancing the light harvesting ability of the polymer. DFT structure optimization further revealed that the more energy-favorable site is C than N for P do**, consistent with the XPS analysis. Moreover, the doped optimized structure enhanced the in-planar distance of nitride pores from 6.95 to 7.04 Å. The electronic structure calculation showed that P do** could able to narrow the band gap from 3.0 to 2.6 eV and the empty midgap states are created by the hybridization of C 2s2p, N 2s2p, and P 3s3p and were well corroborated with UVDRS spectra [196]. Researchers successfully obtained P doped hexagonal tubular g-C3N4 with a hierarchical micro-nanostructure by dissolving melamine and phosphorous acid in deionized water (Fig. 7a) [197]. The hydrothermal process, conducted under pH conditions of 1–3, involved the hydrolysis of melamine into cyanuric acid and forming hexagonal cylinder precursors through self-assembly. The subsequent calcination process allowed the incorporation of P from phosphorous acid into the g-C3N4 structure, resulting in a doped polymer. Scanning electron microscopy (SEM), images revealed the presence of hexagonal rod structures measuring 300–500 nm in length and 60–100 nm in diameter (Fig. 7b). Single-crystal XRD analysis confirmed the presence of multiple H-bonds between melamine and cyanuric acid, arranged in the same plane to form a hexamer topology. The combined hierarchical micro-nanostructure and P do** resulted in a narrowed band gap, increased electrical conductivity, and enhanced photocatalytic properties [198]. Researchers successfully synthesized P doped g-C3N4 nanostructured mesoporous flowers by co-condensing melamine and (hydroxyethylidene)diphosphonic acid in a mixed ethylene glycol solution. A detailed TEM examination revealed the presence of randomly distributed in-plane mesopores on the polymer with sizes ranging from 3 to 18 nm, which correlated well with the surface area analysis. These unique flower-like structures provided abundant active sites, promoting efficient mass transfer and charge separation within nanodomains. The XPS analysis of the P 2p spectrum showed distinct peaks at 131.5 and 133.1 eV, corresponding to P–C and P-N coordination, respectively. This indicated the successful incorporation of P atoms by replacing both C and N atoms in the structure. The introduction of P also induced changes in the structural and electronic properties, evident from the color transformation of the material from pale yellow to dark brown, in agreement with the results obtained from UVDRS. The material exhibited a low recombination rate of charge carriers, as demonstrated by PL and transient photocurrent measurements. Each light excitation triggered rapid, stable, and reversible photocurrent responses. Electrochemical impedance spectroscopy (EIS) revealed a significantly smaller diameter than the bulk polymer, indicating reduced contact and charge-transfer resistance. Furthermore, the material displayed notable cathodic current density and low overpotential, indicating enhanced catalytic activity and reaction kinetics for photocatalytic H2 generation. The flower-like nanostructures with mesoporosity, and P do**, played crucial roles in enhancing light trap**, facilitating mass transfer, and promoting efficient charge separation. These factors contributed to the outstanding catalytic performance of the material compared to the undoped polymer [199]. The doped nanotubes were created through a single-step thermal reaction involving melamine and sodium hypophosphite monohydrate (NaH2PO2·H2O) (Fig. 7c). The formation of nanotube structures was attributed to the in-situ generation and evolution of phosphine gas during the thermal decomposition process. The release of this gas weakened the interlayer van der Waals forces, facilitating the exfoliation of the bulk material into thin layers that then curled up to form nanotubes. At the same time, P was introduced through interstitial do**, which was achieved by reducing uniplanar copolymerization. TEM examination revealed the presence of multilayered nanotubes with a diameter of 200 nm and a thickness ranging from 20 to 50 nm. The P 2p XPS spectra exhibited a peak centered at 133.5 eV, indicating the presence of P in the + 5-oxidation state. The P do** occurred through interstitial incorporation, likely in the form of PO43−, rather than substituting C and/or N atoms. The 31P NMR spectra displayed four distinct signals between 10 and -20 ppm, corresponding to different chemical environments of phosphate radicals within the organic polymeric framework, confirming the findings from XPS analysis. Raman spectroscopy revealed characteristic peaks at 1300 cm−1 (the D band) and 1580 cm−1 (the G band), indicating the presence of surface defects and disorder in the basal plane, respectively. The higher intensity ratio of Id/Ig observed in the doped catalyst suggested an increased presence of defects and disorder. The P do** led to a color change from pale yellow to orange-yellow, resulting in improved absorption of visible light by the material. The VB and CB potentials of the doped catalyst were estimated to be 1.8 and -0.9 eV, respectively, based on VBXPS and bandgap calculations. The decrease in the CB potential from -1.1 to -0.9 eV was consistent with the findings from the Mott-Schottky analysis. The ability to finely tune the conduction potential through do** could affect the selectivity in the photocatalytic reduction of CO2 [200]. A mild and low-temperature method was utilized to create P doped g-C3N4. The process involved a room-temperature mechanochemical reaction between sodium phosphide and cyanuric chloride, followed by annealing at 300 °C for 1 h under argon (Fig. 7d). Through a combination of experimental solid-state analytical techniques and theoretical DFT modeling, the structure was identified as carbon phosphanitride. The P 2p signal analysis revealed a doublet with two peaks at 129.8 and 133.2 eV, indicating P–C coordination and partial oxidation. 31P MASNMR spectra showed two peaks at δP1 = − 12.8 ppm and δP2 = − 24.7 ppm, corresponding to P linked to the triazine-based g-C3N4. Theoretical DFT calculations confirmed the experimental 31P MAS NMR spectra indicating a decrease in the formation of P containing molecules upon annealing [201]. In another study, P doped nanostructured g-C3N4 with an enlarged surface area was produced by incorporating trioctylphosphine oxide (TOPO) as a dopant precursor and SiO2 as a hard template (Fig. 8a). The resulting material exhibited a uniform hollow structure, with the role of TOPO in determining the morphology being crucial (Fig. 8b). The nonpolar characteristics of TOPO, specifically the octyl group, prevented melamine aggregation on the SiO2 surface, leading to the formation of a uniform layer (Fig. 8c). The combination of the hollow structure and P do** demonstrated a synergistic effect, enhancing light absorption across a wide range, promoting charge separation and migration, and increasing the specific surface area. Additionally, the photocurrent showed a 5.4-fold increase in the cathodic direction, and there was a 3.3-fold improvement (Fig. 8d, e) in the current at zero biased potential, significantly enhancing the photoelectrochemical activities [202]. a Under hydrothermal conditions, with the assistance of phosphorous acid at pH levels ranging from 1 to 3, melamine underwent partial in-situ hydrolysis, forming cyanuric acid. The self-assembly between melamine and cyanuric acid led to the formation of regular and stable hexagonal cylinder precursors; simultaneously, phosphorous acid molecules were adsorbed onto the surface of the precursor. Following thermal treatment, phosphorus permeated the framework, forming P doped hexagonal tubular carbon nitride. Reproduced with permission from ref. 197. Copyright (2016) WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. b Large-scale synthesis of P doped g-C3N4 nanotubes was achieved by subjecting a mixture of sodium hypophosphite monohydrate and melamine to high temperatures. The thermal decomposition of NaH2PO2·H2O released phosphine gas, which played a crucial role in transforming the material’s morphology from 2D nanosheets to 1D nanotubes. Reproduced with permission from ref. 200. Copyright (2017) American Chemical Society. c SEM images of P doped g-C3N4. Reproduced with permission from ref. 197. Copyright (2016) WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. d Examples of phosphanitride and phosphorus-containing g-C3N4 were reported by various research groups. Reproduced with permission from ref. 201. Copyright (2022) Wiley–VCH GmbH a The initial step involved the synthesis of SiO2 nanoparticles with an average diameter of 100 nm. Subsequently, P-incorporated g-C3N4 was fabricated using melamine and TOPO through a two-step calcination process. Finally, the removal of SiO2 was achieved using NH4HF2. TOPO played a dual role in the process by acting as a precursor for P do** and as a structure-directing agent, guiding the formation of the desired nanostructured architecture after etching, FETEM images of b h-PCN at 100 nm and c h-PCN at 20 nm scale, LSV curves for the photocatalysts d in the dark and e in illuminated conditions. Reproduced with permission from ref. 202. Copyright (2022) American Chemical Society Researchers successfully incorporated sulfur (S) into g-C3N4 by exposing the polymer powder to a gaseous atmosphere of H2S at a temperature of 450 °C. Analysis using XRD confirmed that the fundamental atomic structure of the material mainly remained intact. However there was a decrease in diffraction intensity, suggesting a reduction in particle size after do**. This finding was further supported by observations using TEM. The reduced particle size played a role in widening the material's bandgap. The S 2p XPS spectrum exhibited two distinct peaks at 163.9 and 168.5 eV, indicating the presence of C-S coordination resulting from the substitution of S for N in the lattice. XANES spectroscopy at the S K-edge revealed two characteristic features at 2472.6 and 2482.0 eV, corresponding to S functional groups: Sθ− (0 < θ < 2) and SO42−. This indicated the presence of S in different chemical environments within the material. DFT calculations were conducted using a 4 × 4 × 2 supercell model, incorporating S dopants that were two-coordinated with N atoms. The calculations demonstrated that the substitutional do** narrowed the bandgap and raised the VB through interactions between S 3p and N 2p orbitals. However, experimental bandgap measurements showed a 0.1 eV increase, contrary to the results of the DFT calculations. This discrepancy was attributed to the influence of quantum confinement effects. The homogeneous substitution of S and the presence of quantum confinement were identified as the factors responsible for the unique electronic structure and enhanced photoreactivity of the doped polymer [203]. S doped nanosheets of two-dimensional g-C3N4 were synthesized by conducting a polycondensation reaction of thiourea followed by a thermal-oxidative treatment that enabled in situ S do**. The thermal oxidative etching process, and S do**, played a crucial role in producing the nanosheet structure, which had an average thickness of around 4.0 nm and increased exposure of S elements on the surface. Examination of the nanosheets using TEM revealed the presence of numerous small surface holes, whose quantity grew with longer etching times, effectively breaking down the layers into nanosheets. The specific surface area, determined through BET analysis, showed a substantial 13-fold increase compared to the bulk material, accompanied by a pore size distribution ranging from 2 to 10 nm. The absorption band edge of the nanosheets exhibited a blue shift from 470 to 420 nm, indicating an increment in the bandgap energy from 2.3 to 2.7 eV. This shift was attributed to the quantum confinement effects resulting from the thermal etching treatment. XPS analysis of the S 2p region peaked at 163.7 eV, indicating successful in situ S do** forming S-C bonds. Moreover, the VB energy increased from 2.2 to 2.3 eV upon S do**. Under irradiation, the nanosheets demonstrated increased transient photocurrents, which returned to zero in the dark. The doped nanosheets exhibited higher photocurrent intensity, suggesting enhanced efficiency in charge carrier separation. First-principles quantum computational studies revealed that S do** shifted the CB edge to more negative potentials than the bulk material, indicating the presence of N defects. Interestingly, S do** slightly shifted the CB edge to a positive potential, similar to the shifts observed in O doped models. These shifts were attributed to the generation of do** levels within the material. Computational calculations further indicated that the most probable surface structures involved the substitution of N with S or O, as they exhibited the lowest formation energy [204]. Fluorination of the polymer was achieved by using ammonium fluoride, which had two effects: it created an open-textured structure and allowed for the adjustment of electronic band gaps. When exposed to light, the fluorinated material exhibited excellent photocatalytic activity. EA confirmed that the C-to-N ratio closely matched the ideal value for g-C3N4 (theoretical C/N ratio of 0.75). XPS analysis detected a peak at 686.2 eV, indicating the presence of C-F bonds. The do** level of F was determined to be 3 atomic%, and solid-state MAS NMR spectroscopy of F (19F) revealed four distinct signals ranging from -60 to -220 ppm, corresponding to different positions within the polymer structure. Incorporating F reduced the band gap from 2.7 eV (for g-C3N4) to 2.6 eV. DFT calculations showed that fluorination played a key role in adjusting the positions of the HOMO and LUMO. The do** process caused both the LUMO and HOMO energy levels to shift upwards. This electronic structure modification through do** significantly impacted the redox properties, thereby enhancing the photocatalytic performance [205]. The co-pyrolysis of melamine and ammonium chloride was employed to demonstrate the incorporation of chlorine (Cl) into g-C3N4. By adjusting the dopant ratio, Cl intercalation was achieved, and XPS analysis of the Cl 2p peak yielded two distinct peaks at 200.4 and 199.1 eV, corresponding to Cl 2p1/2 and Cl 2p3/2, respectively, indicating the successful introduction of Cl atoms into the polymer structure. DFT calculations were utilized to investigate the effects of Cl do** on the polymer and revealed that Cl atoms formed covalent bonds and occupied the interlayers of the polymer, acting as bridges between the layers. This bridged Cl configuration was found to facilitate charge transfer processes and lead to an expansion of the lattice parameters. Furthermore, Cl do** resulted in a narrowing of the band gap, an elevation of both the CB and VB positions, and a potential increase in the polymer's conductivity. The introduction of Cl atoms into the interlayer structure of g-C3N4 resulted a homogeneous porous structure. This led to several advantages, including enhanced charge migration, a higher CB level, a reduced band gap, and an increased surface area. As a result, the Cl doped polymer exhibited excellent photocatalytic performance [206]. Bromine (Br) was used to modify g-C3N4 by mixing urea with different amounts of NH4Br in deionized water at 80 °C. The resulting mixture was sintered in air at 550 °C for 2 h. Raman spectroscopy revealed characteristic shifts in the sample's spectrum, specifically in the 500–1300 cm−1, attributed to heptazine units. These shifts indicated the presence of in-plane bending vibrations of the heptazine linkages (observed at 709 and 762 cm−1) and the symmetric N-breathing mode of heptazine units (observed at 978 cm−1). The absence of peaks in the 2000–2500 cm−1 region indicated the absence of certain linking groups. Solid-state 13C NMR analysis showed two distinct peaks in the spectra of both samples. The stronger peak at 167.4 ppm corresponded to sp2 hybridized C in the form of N–C-N, while the smaller peak at 158.8 ppm was associated with C atoms like CN3 in the melem structure. No new peaks were observed, suggesting the stability of the heptazine g-C3N4 after Br do**. XPS confirmed the presence of Br species with a single peak at 67.8 eV. The introduction of Br into the polymer system resulted in a notable decrease in EIS, indicating improved interfacial charge transfer and migration. Additionally, the doped polymer exhibited higher photocurrent density due to the reduced resistance of the modified material. These findings demonstrate the potential advantages of non-metal elemental do** in modifying the properties of the polymer system [207]. A polymer doped with iodine (I) was synthesized by grinding a mixture of I and bulk material using a planetary ball mill operating at 25 Hz for 50 h. XPS analysis revealed two distinct peaks at 619.3 and 619.9 eV, indicating the presence of C–I and C–I+–C coordination, respectively. The introduction of I caused a noticeable change in the color of the polymer, transitioning it from yellow to a deep brown shade. This color transformation can be attributed to increase delocalization of p-electrons, leading to a shift in the CB value from -1.2 to -1.3 eV [208]. DFT simulations examined the electronic and optical properties of a monolayer g-C3N4 doped with halogens (F, Cl, Br, or I). The researchers discovered that the most stable configuration occurred when the halogen atoms occupied the interstitial space between three tri-s-triazine units within the monolayer. The introduction of halogen atoms led to changes in the DOS and the redistribution of the HOMO and LUMO. Specifically, the highly electronegative F atom tended to occupy the VB and HOMO, while the Cl, Br, and I atoms were involved in the CB and LUMO. The study concluded that do** the monolayer with halogens reduced band gap enhanced light absorption, and decreased work function, which are beneficial for achieving highly efficient photocatalytic activity [209]. Simultaneously incorporating two or more non-metal heteroatoms as dopants in a polymer can lead to significant synergistic effects in catalytic processes [210, 211]. Combining the strengths of multiple dopants make it becomes possible to address various inherent limitations simultaneously, resulting in improved catalytic performance. In some cases, one dopant may modify the material's band structure. At the same time, the other facilitates the incorporation of the active dopant into the host crystal lattice or helps balance charges during aliovalent do** [212,213,214,215]. In other cases, both dopants may work together to finely adjust the semiconductor's band structure cooperatively. When considering substitutional dopants with lower Mulliken electronegativity than C and N (such as B, S, and P), they tend to raise the conduction band minimum (CBM). Conversely, dopants with higher Mulliken electronegativity (such as O, F, and Cl) tend to lower the CBM. Additionally, dopants with more valence electrons than C and N (such as O, S, P, and Cl) tend to create electron-donor states near the CBM. In comparision, those with fewer valence electrons tend to generate electron acceptor states near the VBM [216,217,218,219,220,221,222,223,224]. Overall, co-do** a polymer with multiple non-metal elements has been shown to enhance the catalytic capabilities of the material, leveraging the benefits of single-element do**. g-C3N4 was modified through co-do** with B and S using a thermal co-polymerization method involving melamine, boric acid, and thiourea (Fig. 9a) [225]. XPS analysis revealed distinct peaks in the S 2p spectrum, indicating the presence of various forms of oxidized S. The peak at 162.9 eV indicated the formation of a C-S bond within the aromatic ring. In comparision, peaks at 166.9 and 168.0 eV corresponded to sulfone and sulfate forms of oxidized S (Fig. 9b). The B 1 s peak at 190.5 eV suggested that B replaced C in the aromatic ring, forming a B-N bond. The co-do** of B and S occurred within the lattice of the material. One notable outcome of the co-do** process was the improved ability of the photocatalyst to absorb visible light compared to the undoped material. This enhancement was attributed to a reduced band gap, as evidenced by a shift towards longer wavelengths. The band gap of the co-doped material decreased from 2.7 to 2.5 eV. Additionally, the co-doped photocatalyst exhibited significantly increased current in both cathodic and anodic directions, with an 11- and 8.5-fold increase, respectively, compared to the bulk material. This enhancement in electrochemical performance indicated the positive influence of co-do** on the photocatalytic activity of the material (Fig. 9c). Theoretical investigations using DFT calculations provided insights into the electronic structure changes induced by non-metal do**. In its pure form, g-C3N4 had specific HOMO and LUMO positions. Do** with B resulted in a widened band gap and downward shifts in the HOMO and LUMO positions. Conversely, the introduction of S into the structure led to a shortened band gap and shifts in the HOMO and LUMO positions, reflecting the electron-donating nature of S (Fig. 9d). Simultaneous co-do** with B and S allowed for precise tuning of the HOMO and LUMO positions, taking advantage of the electron-withdrawing and electron-donating properties of the respective dopants. As a result, the HOMO and LUMO positions were adjusted to -5.8 and -1.8 eV, respectively [225]. A new porous nanobelt structure with hierarchical features was successfully synthesized and characterized. This unique material was created using a self-assembly method without templates. The precursor, dicyandiamide, and the dopant source, glutathione, were employed to introduce C and O atoms into the g-C3N4 structure. The resulting co-doped material exhibited a well-defined nanobelt architecture composed of porous nanosheets, significantly increasing its specific surface area to 120 m2 g−1. The material’s bandgap was narrowed by substituting N atoms with C and O heteroatoms allowing, it to harness visible light efficiently. Additionally, the position of the CB shifted downwards, enhancing the material's ability to reduce electrons for H2 production. DFT calculations were employed to determine the precise locations of C and O do** to gain deeper insights. Two potential substitution sites for N atoms, named site 1 and site 2, were carefully examined. The formation energy values were computed for substituting C and O atoms at these specific sites. Notably, substituting an O atom for the N atom at site 1 and a C atom for the N atom at site 2 exhibited significantly lower formation energy values of 0.5 eV and 0.2 eV, respectively. These findings indicate that these specific do** sites are more energetically favorable. By identifying the optimal locations for C and O do**, researchers can finely tune the material's properties to enhance its performance in various applications, including photocatalysis and H2 production [226]. A novel C and O co-doped g-C3N4 material was synthesized using a mechanical mixing technique followed by calcination of hexamethylolmelamine and melamine (Fig. 10a). The characterization analysis confirmed the successful incorporation of C and O into the g-C3N4 structure. The resulting catalyst exhibited two distinct regions with different functions. The C-containing part acted as an electron acceptor, while the O-containing part acted as an electron donor. This donor–acceptor system provided an additional pathway for electron transfer, leading to an expanded optical absorption range and a narrower bandgap of g-C3N4, reducing it from 2.7 to 2.0 eV. Moreover, the donor–acceptor systems enhanced the delocalization ability of photoinduced charge carriers, resulting in prolonged carrier lifetime and reduced electron–hole recombination rates. The improved delocalization of charge carriers can be attributed to efficient electron transfer between the O-containing and C-containing regions of the catalyst. Electrons from the O-containing section readily transferred to the C-containing section, which served as an electron acceptor, establishing an effective electron transfer pathway. This process significantly extended the lifetime of charge carriers and minimized electron–hole recombination. DFT calculations were employed to investigate the mechanism of intramolecular photo-induced electron transfer. Due to the complexity of the catalyst's structure, three representative fragments (F1, F2, F3) were selected for the calculations based on their structural models. The results revealed that the O-containing fragments acted as electron donors, while the C-containing fragments acted as electron acceptors. The cyclic structure of the heterocycle with an O atom exhibited stronger electron donor properties than that of an O atom outside its cyclic structure. Analysis of the spatial separation between the HOMO and LUMO in fragments F1 and F3 demonstrated an efficient mechanism for charge separation (Fig. 10b). In contrast, the incomplete separation in fragment F2 indicated limited charge separation efficiency. The spatial separation of HOMO and LUMO facilitated the separation of charge carriers through the HOMO–LUMO transition, making fragments F1 and F3 ideal for photocatalytic reactions [227]. A new type of g-C3N4 material called P and F co-doped g-C3N4 was synthesized using a sol–gel-mediated thermal condensation process (Fig. 10c). To confirm the chemical structure of the synthesized material, solid-state MAS NMR analysis was conducted. Various MAS and CPMAS NMR spectra, including 13C, 15N, 31P, 19F, and 1H, were obtained for the co-doped material. The 13C CPMAS NMR spectrum revealed the presence of four distinct C peaks, indicating the existence of four different C types in the material. Specifically, two characteristic peaks corresponding to C atoms in g-C3N4 (C3 and C4) were observed at chemical shifts of 160.0 and 168.1 ppm, respectively (Fig. 10d). The synthesized polymer exhibited a well-defined mesoporous nanostructure, which contributed to its improved light absorption properties in the visible and infrared regions. The unique sol–gel synthesis process resulted in a higher BET surface area of 260.93 m2/g, indicating an increased active surface area and potential for high catalytic activity [228]. a A successful synthesis of B and S co-doped g-C3N4 was achieved in a single-step reaction. The procedure involved dissolving melamine, thiourea, and boric acid in water. The solution was then evaporated at 80 °C overnight, forming a white crystalline compound. Subsequently, the compound was subjected to calcination at a temperature of 550 °C for 3 h, b S 2p XPS spectra oxidized sulfur in the form of sulfone and sulfate. c plausible mechanism to explain the photocatalytic reduction of H+ to H2 on the surface of a co-doped catalyst. When exposed to visible light, it undergoes electron–hole pair generation, and the photoinduced electrons in the CB are captured by defects created by introducing of heteroatoms into the carbon nitride lattice. These captured electrons then react with H+ to produce H2 gas with the assistance of Pt nanoparticles. d Atomic orbital compositions of the frontier molecular orbitals and optimized geometries of bulk, S doped, B doped, and B and S co-doped g-C3N4. Reproduced with permission from ref. 225. Copyright (2018) American Chemical Society a 37% aqueous formaldehyde solution and melamine were refluxed in a basic medium to have a clear solution, followed by cooling to room temperature, and a white solid was obtained. The solid and melamine were mixed, and the resulting mixture was subjected to thermal polymerization to obtain C, O co-doped g-C3N4, b DFT optimized fragments (F1, F2, F3) of C, O co-doped g-C3N4 showing O-containing fragments acted as electron donors. In contrast, the C-containing fragments acted as electron acceptors. Reproduced with permission from ref. 227. Copyright (2020) Elsevier B.V. c P, F co-doped g-C3N4 was synthesized by conducting a thermal condensation process using dicyandiamide, [Bmim][PF6], CTAB), and TEOS, during the sol–gel process, TEOS polymerized around CTAB upon thermal treatment. This silica matrix prevented the structural pores’s and trapped PF6 species, enabling their reaction with amino groups of uncondensed carbon nitride precursor. As a result, the PF6 species, became incorporated into the g-C3N4 framework. Subsequent removal of the silica template by washing with ammonium fluoride solution produced silica-free P, F co-doped materials. d Solid-state NMR spectra of the material. Reproduced with permission from ref. 228. Copyright (2020) Wiley–VCH GmbH The controlled generation of vacancies is crucial for harnessing solar energy directly, as it enhances the interaction between defects and photoactivity in photocatalysts. Introducing vacancies, such as C and N, has been shown through theoretical and experimental studies to effectively narrow the bandgap and facilitate the separation of electron–hole pairs. Unlike heteroatom do**, the introduction of vacancies does not introduce impurities into the structure of g-C3N4. By removing graphitic C atoms, the in-plane conjugation system of g-C3N4 is weakened, leading to significant changes in its physicochemical properties. This alteration disrupts the symmetry of g-C3N4, creating electron delocalization effects and defect states that improve the transfer and migration efficiency of photogenerated electrons and carriers. An important benefit of introducing C vacancies is the generation of unsaturated N atoms, which promote catalytic reactions. One effective approach to achieve this is through the thermal treatment of bulk material in an NH3 atmosphere, resulting in holey graphene-like C3N4 nanosheets with abundant in-plane holes. This process increases the exposure of active edges and facilitates cross-plane diffusion channels, leading to accelerated mass and photogenerated charge transfer and reduced aggregation. The resulting polymer exhibits a significantly higher specific surface area of 196 m2/g and an increased bandgap of 2.9 eV. Analysis of the C 1 s spectrum reveals distinct peaks corresponding to C = C and N = C-N bonds, with a higher ratio of N = C-N to C = C peak areas, indicating the removal of graphitic C species through NH3 etching. This reduction in graphitic C content suppresses charge recombination, and the presence of C vacancies enhances the electrical conductivity and mobility of photogenerated charge carriers. XPS analysis shows a significantly lower C/N molar ratio, indicating the ratio modification by C vacancies. This modification extends the light absorption range into the near-infrared region, increases the donor density, and significantly prolongs the lifetime of charge carriers [229]. Researchers have developed a new type of g-C3N4 by intentionally creating vacancies in C atoms through heat treatment in an NH3 atmosphere. These vacancies have significantly improved the performance of the modified polymer, and a detailed analysis has uncovered several factors contributing to this enhancement. High-resolution analysis of the C 1 s spectra revealed two distinct peaks, one corresponding to adventitious C species and the other indicating the presence of sp2 C bonded with N in the aromatic triazine heterocycles. Importantly, the modified sample showed higher binding energy for the latter peak, suggesting a decrease in electron density around the C atoms in the heptazine rings due to the absence of covalently bonded C atoms. Electron spin resonance (ESR) spectroscopy demonstrated a strong ESR signal for the unmodified sample compared to the modified one, which is believed to be caused by local stress and bond-angle disorder in the connectivity structure, with unpaired electrons associated with C atom sites. The introduction of C vacancies has significantly enhanced the polymer's ability to adsorb and activate CO2. Moreover, it has led to an upward shift in the CB, increased charge carrier concentration, and extended lifetime, all of which contribute to improved photocatalytic activity. The study also revealed that C vacancies can mitigate the exciton effect and promote charge carrier generation. This research opens up new possibilities for modifying the exciton effect and charge carrier concentration in the polymer, potentially facilitating the separation of photoinduced electron–hole pairs and enabling charge carrier-involved photocatalytic reactions [230]. The scientists successfully produced and characterized C vacancies in g-C3N4 through calcination in an atmosphere containing H2 gas. This modified form of g-C3N4 exhibits a larger specific surface area and increased active sites due to numerous C vacancies. The material analysis revealed a shift in the position of the N = C = N peak to a higher binding energy, indicating a slight alteration in the arrangement of C atoms. Additionally, the ratio of peak areas corresponding to N = C = N showed changes, indicating the existence of C vacancies resulting from H2 etching. ESR spectroscopy results demonstrated a clear Lorentz line at g = 2.003 for the modified polymers, displaying the strongest response signal. Furthermore, the photocurrent density of the modified material was more than four times greater than that of the bulk material. The C vacancies caused a reduction in electron density around N atoms, narrowing the bandgap and enhancing the material's ability to respond to light. The abundant pore structure and C vacancies facilitated efficient separation and transfer of photo-excited electron–hole pairs, thereby significantly improving the material's photocatalytic efficiency [231]. In contrast, the structure of g-C3N4 remains unchanged when N vacancies are introduced. The study identified the terminal -NHx positions as the most likely locations for creating vacancies among four possible positions (C-N = C, N-(C)3, bridging N, and -NHx). N vacancies generate defect energy levels that can accept electrons and inhibit the recombination of electron–hole pairs, making them valuable as active or adsorption sites. The excess electrons within the N vacancies can react with O2 to produce superoxide radicals or form bonds with metals to create sites for electron absorption. Researchers have employed different techniques, such as N2 or H2 gas calcination, to produce g-C3N4 materials with N defects and achieve high efficiency in photocatalysis. Scientists have developed g-C3N4 nanosheets with controllable N vacancies by subjecting bulk g-C3N4 to an H2 atmosphere at temperatures ranging from 475 to 550 °C for one hour. By analyzing the elemental composition, they observed an increase in the C/N ratio as the heat treatment temperature rose, indicating the formation of N vacancies. XPS analysis confirmed the presence of these vacancies and identified four main components in the N 1 s spectrum. The ratio between specific peak areas decreased with increasing temperature, suggesting the removal of N atoms from certain lattice sites during the thermal reduction process. EPR spectroscopy revealed the presence of unpaired electrons associated with specific C atoms in the material. Both empirical observation and theoretical calculations demonstrated that N vacancies induced midgap states below the CB edge, with deeper states occurring as vacancies increased. The improved photocatalytic activity of the material was attributed to various factors, including enhanced visible light absorption, efficient trap** of photogenerated electrons, and the uniform anchoring of small Pt NPs. However, an excessive induction of N vacancies resulted in deeper midgap states acting as recombination centers, leading to decreased photocatalytic activity [232]. A straightforward and efficient method has been developed to create N-deficient g-C3N4 using alkali-assisted thermal polymerization of urea, melamine, or thiourea. The introduction of N vacancies has a significant impact on the optical properties of the material, causing a shift in the absorption edge towards the red region of the spectrum. The concentration of defects can be controlled by adjusting alkali to N precursor ratio. Under visible light, the N-deficient samples exhibit superior photocatalytic performance compared to the pristine samples. Solid-state 13C MAS NMR measurements confirm the presence of newly-formed cyano groups, with distinct peaks observed in the spectra. The introduction of N defects does not significantly affect the VB position, but it does lead to a decrease in the CB position, resulting in a narrower bandgap. DFT calculations further support these findings by demonstrating a decrease in the bandgap energy when cyano groups and N vacancies are present. The narrowing of the bandgap can be attributed to the coexistence of cyano groups and N vacancies in the material [233]. A novel technique was developed to create porous g-C3N4 materials with many defects. This was achieved through a short thermal treatment in an air environment without additional reactants. The resulting materials demonstrated an enhanced ability to absorb light in the visible spectrum. The red-shifted absorption edge was attributed to the presence of cyano groups, while the appearance of a new absorption edge was linked to N vacancies. Theoretical calculations of the dielectric function and previous research findings supported these observations. It was proposed that including cyano groups and N vacancies within the structure of g-C3N4 led to a reduction in the bandgap and the introduction of energy levels associated with defects, resulting in an expanded range of light absorption [234]. A groundbreaking method was developed to produce defect-modified, thin-layered, and porous g-C3N4 materials using urea- and KOH-assisted thermal polymerization. This technique simultaneously introduces cyano groups, N vacancies, and mesopores into the g-C3N4 structure while reducing its thickness. By adjusting the mass ratio of urea, KOH, and melamine, the thickness, pore characteristics, and defects can be finely tuned, resulting in a significant increase in specific surface area, light absorption capacity, and the efficiency of separating photoexcited charge carriers. These advancements greatly enhance mass transfer within the g-C3N4 material. Solid-state and 13C CPMAS NMR measurements were conducted to analyze the samples. The spectra showed distinctive features, with prominent peaks at 156.3 and 164.5 ppm representing the chemical shifts of C3N atoms and C2N–NHx atoms in heptazine. The peak intensity at 156.3 ppm decreased with higher KOH usage, resulting in a reduced intensity ratio of C3N to C2N–NHx from 0.29 to 0.14. New peaks at 123.3 and 171.4 ppm were also observed, corresponding to the C atoms in cyano groups and neighboring C atoms, respectively. These findings suggest that the introduced cyano groups are likely located at the apex of the melon structure, leading to partial fracturing and restructuring of N = C–N2 units and a decreased concentration of C3N atoms. Orbital analyses of both pristine and modified samples were conducted, revealing significant differences. In the pristine sample, the charge density of the LUMO and the HOMO was uniformly distributed over the heptazine rings. In contrast, the modified sample showed redistributed charge density, resulting in the emergence of electron-rich regions and well-separated distributions corresponding to LUMO and HOMO. These changes can be attributed to the increased local fluctuations of the electrostatic potential caused by the introduced N defects. The distinct and localized charge density distribution facilitates efficient separation and transfer of photoexcited charge carriers, minimizing back reactions and significantly enhancing the photocatalytic activity [235]. The physical, chemical, and optoelectronic properties of g-C3N4 can be finely tuned through thermal treatment, which introduces N vacancies and defects/holes in the nanosheets, increasing porosity. These modifications significantly impact the material's optoelectronic characteristics, including the appearance of n → π* transitions, a redshift in light absorption, enhanced charge separation, and reduced charge recombination. Trap states near the VB further contribute to improved charge separation. Analyzing the C and N core levels provides valuable insights into the material's composition. The C core level exhibits three peaks, with the main peak attributed to sp2 hybridized C in the tri-s-triazine rings, while the weaker peaks at 285.7 and 288.7 eV originate from surface oxidation of the C film. The N core level can be deconvoluted into three peaks representing sp2 hybridized aromatic N atoms in C–N = C bonds, sp3 hybridized N atoms in N-(C)3 groups, and terminal amino groups (C–NH2 or = NH), respectively. Analysis of the N content reveals an increased atomic ratio of C/N from 0.73 to 0.76, indicating a relative enrichment of C in the samples due to the presence of N vacancies in the g-C3N4 structure [236]. Controlling the morphology and surface vacancies of g-C3N4 is crucial for enhancing the efficiency of photocatalytic systems. A groundbreaking study demonstrated the positive impact of N vacancies in the atomic layers of g-C3N4 on the photoreduction of CO2. The researchers achieved a unique 3D bubbly structure of g-C3N4 nanosheets, which disrupted the interactions between adjacent layers and facilitated charge transport. They used an NH4Cl gas template that decomposed upon heating, resulting in a bubble film on the nanosheet surface. The researchers could tune the N vacancies by intentionally introducing N defects into the ultrathin nanosheets under a reducing atmosphere. This N vacancy tuning significantly affected the optoelectronic properties of the nanosheets. It increased the reducing ability of the nanosheets, extended their light absorption range to longer-wavelength visible light, and prolonged the radiative recombination of electron–hole pairs. These improvements were attributed to midgap states, which efficiently trapped electrons and acted as reservoirs [237]. When metal NPs are placed on the surface of a semiconductor, there is a difference in their work functions that creates a contact potential difference known as the Schottky barrier [238, 239]. This leads to bending of the energy bands at the interface once equilibrium is reached. The band bending extent depends on the metal and semiconductor's relative work function energies. This phenomenon promotes the directional movement of photo-generated electrons from the semiconductor to the metal, significantly improving the separation of charges. As a result, the system effectively traps electrons, reducing recombination between electron–hole pairs [240,241,242]. The combination of plasmonic [243,Full size image g-C3N4 nanosheet panels decorated with Au NPs oriented with (111) facets were synthesized using a two-step method involving vapor-deposition polymerization and immersion (Fig. 11d). Characterization using TEM and AFM revealed that the film consisted of g-C3N4 nanoplatelets with a thickness of 12 nm and equally distributed Au NPs with a diameter of approximately 2 nm (Fig. 11e). The resulting photocatalyst exhibited enhanced absorption of visible light due to the surface plasmon resonance effect of the Au NPs. This, along with efficient transportation and separation of charge carriers (Fig. 11f) and well-aligned VB levels, enabled effective overall water splitting [268] without the need for sacrificial agents. The photocatalyst demonstrated stoichiometric evolution of H2 and O2, distinguishing it from the half-reaction observed when using Pt-decorated g-C3N4. Amine-functionalized g-C3N4 was treated with N2 plasma and impregnated with HAuCl4.3H2O to create Au NPs. These NPs were then deposited onto g-C3N4 nanosheets. Notably, g-C3N4/Au exhibited enhanced photocatalytic activity for reducing CO2 under visible light without additional cocatalysts or sacrificial agents. The rates of CO and CH4 evolution achieved by g-C3N4/Au were significantly higher compared to pristine g-C3N4, with values of 28.3 and 1.3 μmol h−1 g−1 respectively, and these rates were 7.6 and 2.6 times higher than bulk [269]. The improved performance of g-C3N4/Au can be attributed to several factors. Firstly, introducing amine groups enhanced the adsorption capacity of g-C3N4/Au for CO2. Secondly, small-sized Au NPs (Fig. 12a) generated hot electrons that activated surrounding electrons through energy transfer, resulting in increased local temperature and improved efficiency in the CO2 reduction reaction. Finally, forming a Schottky junction between Au and g-C3N4 facilitated electron migration from g-C3N4 to Au NPs (Fig. 12b), effectively reducing carrier recombination. The catalytic activity of metal–semiconductor heterojunctions in CO2 conversion relies on precisely tuning the Schottky barrier. Researchers successfully demonstrated the utilization of plasmonic Ag NPs on N-vacancy modified g-C3N4 nanotubes through a simple self-assembly method (Fig. 12c), enabling efficient visible-light photocatalytic CO2 conversion. The figures revealed that highly dispersed small Ag NPs, ranging from 6 to 8 nm, were uniformly loaded onto the surfaces of nanotubes (Fig. 12d). This indicated that the one-dimensional g-C3N4 nanotubes, enriched with reductive active sites, greatly improved CO2 transmission/adsorption, captured photo-generated electrons, and established a desirable Schottky heterojunction for effective separation and migration of charge carriers. The homogeneous distribution of Ag NPs and the directed spatial arrangement within the 1D tubular g-C3N4 structure significantly enhanced the utilization efficiency of plasmonic metals. The nanocatalyst exhibited an impressive CO evolution rate of 88.2 μmol g−1 h−1 under visible light. The optimized Schottky barrier height not only facilitated charge kinetics through the driving force from the Schottky junction but also prevented the trapped photoelectrons from flowing back to g-C3N4 under visible light. This effectively suppressed the recombination of photoinduced charge carriers, resulting in highly efficient CO2 photoreduction. The favorable spatial distribution and precisely tuned Schottky barrier in the nanotube heterojunction contributed to its superior photocatalytic performance [270]. a TEM and HRTEM images of g-C3N4/Au. b The proposed mechanism for the photocatalytic reduction of CO2 by g-C3N4/Au involves the synergistic effects of introduced amino groups, activation of hot electrons, and formation of a Schottky junction. These factors work together to enhance the photocatalytic activity of g-C3N4/Au, enabling the efficient conversion of CO2 into valuable products. Reproduced with permission from ref. 269. Copyright (2020) Elsevier Inc. c The self-assembly of melamine and cyanuric acid, resulting in the formation of well-defined 1D pillar supramolecular precursors, which can connect to through hydrogen bonding within the layers and grow vertically in adjacent interlayers through π-π interactions, subsequently, Ag(I) ions are introduced into the supramolecular precursors using AgNO3. During the thermal polycondensation process, the combined effects of high temperature and gas release promote the exfoliation of the supramolecular pillars into ultra-thin layers. Each 1D hexagonal pillar contains tightly packed nanosheet-like frameworks within its interior, and to minimize surface energy, the exfoliated nanosheets gradually curl up and transform into g-C3N4 nanotubes. d TEM image of g-C3N4 nanotube with well-distributed Ag NPs. Reproduced with permission from ref. 270. Copyright (2022) Elsevier B.V.2 History of graphitic carbon nitride

4.1 Single non-metal do**





4.2 Bi-non-metal do**


4.3 Vacancy creation
4.4 Plasmonic metal loading

5 Photocatalytic application
This section focuses on the progress of photochemical processes involving photocatalysis, specifically the splitting of water to produce H2 and O2, as well as the reduction of CO2 to hydrocarbon fuels. It is important to note that these applications have been extensively explored and discussed in various review articles. However, the goal is to provide readers with a concise yet comprehensive overview of the potential benefits of doped g-C3N4-based photocatalysts in these applications. A collection of examples based on recent research findings is presented without favoring any specific research group.
5.1 Water splitting reaction
Using a photocatalyst and solar energy to produce H2 gas from water is a promising avenue in the quest for renewable energy sources, offering a viable alternative to traditional fossil fuels. H2 is renowned for its exceptional energy density, surpassing standard hydrocarbon fuels such as gasoline and diesel. Recent advancements in doped nanomaterials based on g-C3N4 have led to highly efficient photocatalytic water-splitting processes (Table 1). These processes involve the converting light energy into H2 and O2 gases, with photogenerated electron–hole pairs migrating to active sites on the surface of the g-C3N4 photocatalyst. At these sites, they facilitate the reduction and oxidation reactions of water molecules, resulting in the generation of gaseous H2 and O2 without recombination, as depicted by Eqs. 1–3.
Researchers led by Wang conducted a study where they synthesized g-C3N4 by subjecting cyanamide to high temperatures for 4 h. They then utilized this material in the photocatalytic water, splitting it into H2 and O2 using visible light. However, the original g-C3N4 showed inconsistent rates of H2 evolution, ranging from 0.1–4 μmolh−1, with significant variation between different batches. To address this issue, they modified the g-C3N4 by adding 3wt% Pt, resulting in a remarkable increase in H2 production. After 72 h, they could generate 770 μmol of H2, surpassing the amounts produced by both C6N8 units and Pt NPs. For the production of O2, they introduced RuO2, a common cocatalyst for O2 generation, along with AgNO3 as a sacrificial electron acceptor. Under ultraviolet illumination for 8 h, they achieved a total O2 evolution of 53 μmol. In their quest to enhance photocatalytic performance, the researchers explored rational design strategies as a promising alternative. They focused on engineering O doped g-C3N4 nanotubes, which exhibited an impressive H2 evolution rate of 73.84 μmolh−1. This surpassed the performance of previously reported one-dimensional g-C3N4 photocatalysts. The exceptional performance of the nanotubes was attributed to the deliberate incorporation of O, which improved the band structure of g-C3N4. This resulted in a reduced recombination rate of photogenerated carriers and a narrower band gap. Moreover, the unique hollow nanotube structure of the O-doped g-C3N4 facilitated multiple diffuse reflections, thereby enhancing the utilization of visible light and further enhancing the efficiency of the photocatalytic water-splitting process [277]. Phosphorization of g-C3N4 resulted in an improved ability to generate H2 through photocatalysis. This enhancement can be attributed to several factors, such as a narrower bandgap, a higher CB edge, better separation and transfer of photogenerated charges, and a highly hydrophilic surface that promotes the absorption of reactant molecules during the photocatalytic process. Notably, the modified catalyst exhibited an H2 production rate twice as high as the pristine g-C3N4, reaching 916.2 μmolg−1 h−1, with an apparent quantum yield (AQY) of 6.52% at 420 nm. Additionally, the modified catalyst achieved an AQY of 1.48% at 500 nm and 0.7% at 520 nm, indicating that the expanded light absorption beyond 500 nm contributed to its significantly increased H2 production rate [317]. The researchers investigated the photocatalytic H2 production capability of S doped holey g-C3N4 nanosheets under visible light. The nanosheets demonstrated impressive activity, with an H2 evolution rate of 6225.4 μmolg−1 h−1 and an AQY of 10% at 420 nm. Through extensive characterization and theoretical calculations, it was determined that the outstanding performance of the nanosheets can be attributed to their distinct properties. These properties include an increased surface area, a downward shift of the CB, and a reduced bandgap resulting from S do** and the unique ultra-thin two-dimensional structure of the nanosheets. These factors synergistically contribute to the enhanced photocatalytic performance of the material [317]. Utilizing a Schiff-base reaction that involves substituting an N atom with a C atom, the bandgap of g-C3N4 was effectively reduced from 2.7 to 2.6 eV. This narrowing of the bandgap facilitated a more efficient dissociation of photogenerated excitons. The resulting C self-doped g-C3N4 exhibited remarkable rates of photocatalytic H2 evolution, surpassing those of pure g-C3N4 when exposed to both blue and visible light. The optimized sample demonstrated excellent performance with H2 evolution rates of 94.0 and 28.4 μmolg−1 h−1, even under green and yellow light illumination, respectively. These findings highlight the versatility of this approach in enhancing the photocatalytic performance of g-C3N4 [275]. In a recent study, researchers evaluated the effectiveness of ultrathin g-C3N4 doped with N and O for photocatalytic H2 production. They used a Pt co-catalyst and observed a significantly higher H2 evolution rate than previous studies on 2D g-C3N4 nanostructures (Fig. 13a). Increasing the Pt loading (Fig. 13b) further enhanced the H2 production. The study confirmed that the photocatalytic activity was driven by photogenerated electrons, with an AQY of 16.2% under irradiation at 405 ± 10 nm. The researchers also noted the high stability of the photocatalyst under the experimental conditions. They observed consistent and reproducible high activity without any noticeable decrease in the H2 production rate after multiple cycles within 18-h period. Characterization techniques were employed to analyze the distribution of Pt, revealing its presence in clustered form. Additionally, the study investigated the influence of irradiation wavelength on the photocatalytic H2 production activity. The observed variations in activity aligned with the shape of the UV–visible absorption spectrum [293]. The research investigated the photocatalytic H2 evolution capabilities of a nanomesh composed of B and O co-doped g-C3N4 under visible light exposure, utilizing a sacrificial agent, Triethanolamine (TEOA), and Pt cocatalyst. The results revealed that the O doped g-C3N4 nanomesh exhibited an H2 evolution rate approximately 12.7 times higher (345 μmolh−1 g−1) than that of bulk g-C3N4, thanks to its porous nanostructure and O do**. Interestingly, the B and O co-doped g-C3N4 nanomesh demonstrated even higher H2 evolution rates (9751 μmolh−1 g−1) than the O doped version, indicating a significant enhancement in photocatalytic performance due to B do**. The stability of the B and O co-doped g-C3N4 nanomesh was also assessed and confirmed through XRD and SEM analysis, demonstrating its structural integrity during the photocatalytic H2 production process [290]. In a separate scientific investigation, introducing P and O into g-C3N4 significantly improved its photocatalytic performance. The doped material exhibited an H2 production rate of 1588 μmolh−1 g−1, which was approximately double the rate achieved by pristine g-C3N4 (Fig. 13c). Furthermore, the P and O co-doped g-C3N4 displayed excellent stability and the ability to be reused for up to 12 h without a noticeable decline in its photocatalytic activity. This suggests that the doped g-C3N4 has the potential to serve as an efficient and reliable photocatalyst for H2 generation [318]. Adding N defects to g-C3N4 has shown promise in boosting the photocatalytic performance of g-C3N4-based materials for H2 production. N defects can alter the material's electronic structure, creating active sites facilitating catalytic reactions. This modification enhances the material's ability to absorb visible light, essential for efficient H2 production through photocatalysis. Introducing N defects involved a controlled treatment of pure g-C3N4 with selenium vapor. The resulting N-defected g-C3N4 exhibited an average H2 generation rate of 1.16 mmolg−1 h−1 at room temperature (25 °C), which is 3.4 times higher than that of pristine g-C3N4. These findings highlight the significant improvement in photocatalytic activity achieved by incorporating N defects into g-C3N4 [319]. A novel and cost-effective method was used to prepare a 3D porous polymer with a high surface area and N defects without needing templates. This modified material, characterized by its nanocage-like structure, demonstrated enhanced photocatalytic performance compared to pristine g-C3N4. The N defects and nanocage-like structure synergistically contributed to a wider range of responsiveness to visible light, increased exposure of active sites, and improved separation of photogenerated carriers. As a result, the modified g-C3N4 exhibited remarkable performance in the H2 evolution reaction during water splitting under visible light, achieving a maximum H2 evolution rate of 92.6 μmolh−1. This rate was approximately 20.3 times higher than that of pristine g-C3N4, showcasing the significant improvement achieved by introducing of N defects [311]. A surface hydrotreating method was employed to address the issue of unreacted amino groups hindering the photocatalytic activity of bulk g-C3N4. By treating the bulk g-C3N4 with NaBH4 under an inert atmosphere, hydrogenated deficient g-C3N4 was produced. This process resulted in the complete reduction of unreacted amino groups (-NHx) and the formation of N vacancies and cyano groups (-C≡N groups) within the g-C3N4 structure. This elimination of -NHx groups effectively mitigated photo-induced carrier recombination, significantly improving photocatalytic H2 evolution. The H2 production rate achieved with the modified material reached 340.6 μmolh−1, 2.5 times higher than the bulk g-C3N4. Furthermore, the modified material exhibited an AQY value of 16.2% at 420 nm, indicating its enhanced ability to convert light energy into H2 [320]. A simple and efficient method involving the thermal polymerization of urea and KOH has been developed to create defect-modified thin-layered and porous g-C3N4. This process reduces the thickness of g-C3N4 and introduces cyano groups, N vacancies, and mesopores simultaneously. These modifications greatly enhance the photocatalytic efficiency of g-C3N4 for H2 evolution. When exposed to light with a wavelength greater than 420 nm, the defect-modified g-C3N4 demonstrated an H2 evolution rate of 1.5 mmolh–1 g–1, more than 48.5 times higher than that of pristine g-C3N4. Moreover, the defect-modified g-C3N4 exhibited an impressive average AQY of 18.5% at 500 nm, indicating its superior ability to convert light energy into H2 [235]. A simple three-step heat-treatment method is employed to create defective ultrathin mesoporous g-C3N4. This method brings the photogenerated carriers closer to the surface, enhancing the adsorption and diffusion of reactants and products while exposing more active surface sites. Additionally, the high-temperature hydrogenation process generates N-vacancy defects that improve light absorption, boost intrinsic carrier mobility, and increase the availability of active sites. Consequently, the photocatalyst demonstrates an impressive rate of photocatalytic H2 production, reaching up to 13.6 mmolh−1 g−1 under visible light in a TEOA solution, as well as achieving a rate of 33.5 μmolh−1 g−1 for overall water splitting [295]. A novel method has been developed to create thin polymeric g-C3N4 nanosheets with controlled surface C vacancies, greatly enhancing their ability to produce H2 through photocatalysis. These nanosheets have unique structures with improved surface properties, resulting in a higher number of active sites and a significantly increased rate of H2 evolution compared to bulk material. Furthermore, these nanosheets exhibit remarkable stability under repeated testing, showing no significant decrease in activity even after undergoing four cycles. At a wavelength of 405 nm, the nanosheets achieve an AQY of 11.3%, indicating their potential to improve the efficiency and stability of photocatalytic H2 production significantly [321]. A novel fibrous g-C3N4 material was developed using a unique method involving pyrolyzing functionalized 2,5-thiophenedicarboxylic acid and melamine precursors in a laminated covalent triazine framework. C vacancies were created by subjecting the material to a two-step calcination process in argon and air atmospheres, exposing the nanofibers’ edges and diffusion channels. This resulted in the formation of additional active sites and facilitated the transfer of photogenerated charges. The synthesized material exhibited remarkable stability and achieved a maximum H2 evolution rate of 102.6 μmol in 6 h under visible-light irradiation, surpassing the H2 production rate of the bulk material by 66 times. Furthermore, the fibrous g-C3N4 maintained its stability even after undergoing ten cycles of visible-light irradiation [322].

a Under visible light irradiation, the N, O co-doped g-C3N4 nanosheets demonstrated continuous and stable hydrogen production for 6 h during water splitting. The average rate of hydrogen evolution was significantly higher than that of previously reported 2D g-C3N4 nanostructures. b Different Pt loadings as a cocatalyst influence the hydrogen evolution process. Reproduced with permission from ref. 293. Copyright (2019) WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. c The rate of H2 evolution shows improvement as the mass fraction of ammonium polyphosphate increases from 0 to 10wt% during fabrication. However, if the ammonium polyphosphate content is further increased beyond this range, the H2 evolution rate decreases in the case of P, O co-doped g-C3N4. Photocatalytic reduction activity for CO2 conversion, formation of intermediate CO, and final product CH4 on P doped g-C3N4 exhibited a linear increase as the irradiation time progressed. Reproduced with permission from ref. 318. Copyright (2022) American Chemical Society. Yield of (d) CO and (e) CH4. Reproduced with permission from ref. 200. Copyright (2017) American Chemical Society. f CO evolution on the g-C3N4 and carbon vacancy modified g-C3N4 photocatalysts over 5 h. Reproduced with permission from ref. 230. Copyright (2019) The Royal Society of Chemistry
A thermal treatment method has been developed to enhance the photocatalytic activity of g-C3N4 for water oxidation by introducing B dopants and N defects simultaneously. This treatment modifies the material's band structure and electronic properties, significantly improving in the rate at which O2 is evolved. By adjusting the positions of the CB and VB, the material becomes more efficient in absorbing visible light and driving the water oxidation process. The optimized O2 evolution rate of the modified g-C3N4 is approximately six times higher than that of the pristine material, reaching 561.2 µmolh−1 g−1 compared to 98.6 µmolh−1 g−1 [182]. A nanostructured photocatalyst based on g-C3N4 was developed, incorporating P to enhance its performance; including P in the g-C3N4 framework increased surface area, leading to improved light absorption across a wide range of wavelengths. This modification enhanced the separation and migration of charges, and raised the photocatalyst’s specific surface area. As a result, the photocatalyst demonstrated impressive overall water splitting activity, achieving a high faradic efficiency of 95%. Without needing additional cocatalyst or sacrificial agent, the photocatalyst produced an H2 evolution rate of 27 μmolh–1 and an O2 evolution rate of 13 μmol h–1 [202].
5.2 Carbon dioxide reduction
With growing global concerns about climate change and the escalating levels of CO2, there is a pressing need to find efficient methods for utilizing solar energy to convert CO2 into valuable fuels and products. One way to enhance the photo-assisted selective conversion of CO2 is by using g-C3N4 with appropriate reduction potential values and incorporating foreign elements. The process of visible light-driven photocatalytic CO2 conversion involves several steps. These steps encompass the adsorption of CO2 molecules and reductants onto the photocatalyst's surface, the generation of photocarriers upon absorption of visible light, the migration of photo-illuminated excitons across the semiconductor material's surface, surface photocatalytic reactions between the adsorbents and photocarriers, and the subsequent desorption of photo-reaction products, followed by the re-adsorption of reactants. the supplied potential must be more negative than the required standard reduction potential to enable the spontaneous conversion of CO2 (ΔG < 0). Equations (4) to (10) provide the formal redox potential values (relative to NHE at pH 7) that facilitate the photocatalytic reduction of CO2 reactions.
The CO2 adsorption capacity of pristine g-C3N4 is limited, which hampers its effectiveness in catalyzing CO2 reduction. However, researchers have explored methods to enhance the adsorption capacity by introducing vacancies or dopants into the material. In a study [230], the concentration of C vacancies in g-C3N4 was directly related to its ability to adsorb CO2. Linear CO2 molecules have low reactivity and pose a challenge for photocatalytic reactions. A critical mechanism for CO2 reduction involves activating adsorbed CO2 through single-electron transfer at active sites, resulting in the formation of CO2•− intermediates. However, this step is thermodynamically unfavorable due to the high LUMO level of CO2, leading to a highly negative equilibrium potential. The transformation of linear CO2 to bent CO2•− radicals requires significant energy rearrangement, making the single-electron activation step the limiting factor in CO2 photoreduction. Nonetheless, the presence of defects in g-C3N4 facilitates CO2 activation by enhancing its adsorption. These defects, including vacancies and dopants, play a crucial role in promoting the overall process of CO2 photoreduction (Table 2).
A comprehensive approach that combines experimental investigations and theoretical calculations is essential to gain a deeper understanding of the CO2 reduction reaction (CO2RR). DFT has proven to be particularly crucial among the various computational methods available. The fundamental principles of DFT were initially proposed by Hohenberg and Kohn in 1964 [342]; DFT has been pivotal in providing valuable insights into the fundamental properties of systems with multiple particles. It has contributed to understanding of various aspects of CO2 reduction reactions, such as electronic excitations, energy band structures, adsorption energies, and defect sites. Moreover, DFT calculations can explore potential reaction pathways by analyzing the elementary steps involved. DFT calculations have played a crucial role in understanding how the structure of g-C3N4 relates to its catalytic performance. In a study led by Su and their research group, DFT was used to investigate the mechanisms behind the improved performance of S doped g-C3N4. The calculations revealed that S atoms can be doped into three possible sites: NCen, NAro, and NTet. However, NTet sites were considered insignificant and excluded from further analysis. By examining the formation energies (Eform) of S doped g-C3N4, the researchers determined that do** at the NAro site was the most stable, with Eform values of -1.6 eV for NCen and -3.0 eV for NAro. The DFT calculations also provided insights into the band structure and partial density of states (PDOS). S doped g-C3N4 exhibited a smaller band gap (2.4 eV) than pristine g-C3N4 (2.7 eV), allowing for better absorption of visible light. Analysis of the PDOS revealed that the VB of g-C3N4 primarily consisted of N atoms, while the CB was predominantly composed of C atoms. In contrast, the VB of S doped g-C3N4 involved C, N, and S atoms, while the CB contained C and N atoms. Significantly, S do** caused a shift in the Fermi level towards the CB, indicating an increased reduction ability in S doped g-C3N4. The calculated LUMO and HOMO supported the altered orbital distributions in S doped g-C3N4, facilitating efficient separation of electron–hole pairs generated by light and enhancing the photocatalytic efficiency of the material [343]. DFT calculations are also essential for examining the effects of N vacancies on material properties. For instance, when comparing the PDOS of g-C3N4 nanosheets with N vacancies to that of bulk g-C3N4, it becomes evident that the N vacancies introduce new states in the middle of the energy gap near the bottom of the CB. This modification enables more efficient excitation of electrons during the photoreduction process, enhancing the material's ability to undergo photo-induced electron transfer [232]. In addition, DFT calculations play a crucial role in investigating the pathways involved in the reduction of CO2 on different catalyst surfaces. For instance, Du et al. conducted DFT calculations to examine the reaction barriers of CO2 reduction on Pd/g-C3N4, explicitly focusing on forming formic acid (HCOOH) and methanol (CH3OH). The study identified the hydrogenation of CH2OH* and HCOO* as the rate-determining steps for the CH3OH and formaldehyde (HCHO) pathways, respectively. The calculations revealed that Pd/g-C3N4 prefer to produce HCOOH due to the lower energy barrier associated with the HCOOH pathway and the favorable desorption of HCOOH [344].
S, P, B, O, and halogens are non-metallic elements used as dopants to enhance the photocatalytic performance of g-C3N4 in CO2 reduction. Among these dopants, S do** has been extensively studied for its ability to improve the photoreactivity of g-C3N4. Wang and colleagues proposed a simple method to prepare S doped g-C3N4 by thermally treating a thiourea precursor, while undoped g-C3N4 was obtained from melamine. XPS analysis confirmed the formation of C-S and N-S bonds from S do**. S doped g-C3N4 showed a shift in absorption wavelength towards longer wavelengths and exhibited increased absorption in the ultraviolet region compared to the undoped material. This suggests that S do** enhances light utilization, generating photo-induced electrons and holes more. DOS calculations for S doped g-C3N4 and pure g-C3N4 revealed that the CB and VB mainly comprise C 2p and N 2p states. In pure g-C3N4, the bandgap between the VB and CB aligns with the maximum energy band. However, in S doped g-C3N4, an intermediate energy level consisting of C 2p, N 2p, and S 3p states appears below the CB, decreasing the maximum energy band. This modification significantly improves the transition probability of photo-induced electrons. Additionally, S do** introduces more defects in g-C3N4, which facilitates charge transfer and separation and extends the lifetime of charge carriers. Under UV–visible light irradiation for 3 h, S doped g-C3N4 demonstrated a higher yield of CH3OH production compared to the bulk material. However, the effect of S content on the activity and product selectivity in CO2 reduction has not been thoroughly investigated. Therefore, S do** shows potential for enhancing CH3OH yield and utilizing cost-effective rare-earth elements (S, P, N) to improve CO2 reduction activity and selectivity [331]. P doped g-C3N4 was synthesized using a simple poly-condensation method involving melamine and NaH2PO2·H2O precursors. During the formation process, phosphine gas released from the decomposition of NaH2PO2·H2O caused the ultrathin layers of g-C3N4 to roll up, forming nanotubes instead of the usual sheet-like structure. This unique nanotubular structure provided larger surface areas and increased the number of surface sites enriched with amino groups. Moreover, P do** shifted the absorption edge of g-C3N4 towards longer wavelengths, indicating enhanced absorption of visible light. When exposed to visible light for 4 h, the P doped g-C3N4 exhibited significantly higher yields of carbon monoxide (CO) and methane (CH4), reaching 9.5 and 7.2 μmolg−1, respectively (Fig. 13d, e). These values were 3.1 and 13.9 times higher than those observed with pure g-C3N4. Additionally, the reduced CO/CH4 ratio from 6.0 to 1.3 indicated that P do** selectively promoted the production of CH4 [200]. In a study conducted by Yu and colleagues, they investigated the incorporation of O dopant into g-C3N4. Their research revealed that O atoms preferentially replaced bi-coordinated N atoms in the aromatic heterocycles of g-C3N4. Through a thermal delamination and curling-condensation process under high-temperature oxidation conditions with air flow, they successfully synthesized hierarchical porous nanotubes of O doped g-C3N4. The catalyst exhibited a significantly larger specific surface area of 36 m2g−1 compared to the 9 m2g−1 surface area of bulk g-C3N4. This increase in surface area provided more active sites for CO2 adsorption and facilitated efficient charge transfer. The introduction of O dopants also resulted in a shift in the bandgap, enhancing its light absorption capacity and generating a greater number of photo-induced charge carriers. Notably, it demonstrated an average CH3OH generation rate of 0.9 μmolg−1 h−1, nearly five times higher than that observed for pristine g-C3N4 [345]. A novel technique involving sol gel-mediated thermal condensation of dicyandiamide has been successfully employed to synthesize amorphous g-C3N4 doped with P and F. Introducing P dopants creates midgap states below the CB, formed through the hybridization of C, N, and P orbitals. This reduces the bandgap, indicating transitions between the VB and midgap states or from midgap states to the CB. The specific position of the midgap states in the co-doped catalyst surpasses the CO2/CH3OH potential, meeting the thermodynamic requirements for CO2 photoreduction. The researchers achieved a high CH3OH yield, surpassing previous studies on g-C3N4 catalysts doped with heteroatoms or undoped for the photocatalytic conversion of CO2 to CH3OH. The improved photocatalytic activity of the co-doped g-C3N4 is attributed to the reduced bandgap and increased presence of structural defects. The narrower bandgap allows for enhanced light absorption, generating a larger number of electrons upon irradiation. The incorporation of F modifies the electronic structure of the g-C3N4 network, narrowing the bandgap and promoting p-π conjugation, thereby facilitating the transfer of photocarriers. Defects in the co-doped catalyst suppress the recombination of charge carriers. It extends the lifetime of electrons, leading to more available charge carriers for CO2 reduction. The co-doped g-C3N4 efficiently absorbs visible light, leading to the generation of electron–hole pairs. The holes then oxidize water molecules, producing protons and hydroxide radicals, which further react to generate O2 and additional protons. These protons and CO2 are adsorbed on the catalyst surface, and the photo-generated electrons reduce them to produce CH3OH. By aligning the CB with the redox potential of CO2/CH3OH, the formation of unwanted byproducts such as HCOOH and HCHO is minimized [228].
A new method was employed to synthesize co-doped g-C3N4 using a sol gel-mediated thermal condensation technique with dicyandiamide as the precursor. The co-do** involved incorporating both C and O into the g-C3N4 structure. The co-doped catalyst demonstrated CO as the primary product, accompanied by a small quantity of CH4. When exposed to UV light, the pure g-C3N4 catalyst exhibited a CO evolution rate of 8.11 μmolg−1 over a 4-h duration. However, introducing hexamethylenetetramine (HMT) do**, significantly increased the CO evolution rate, reaching 4.3 times higher than pure g-C3N4. It is worth noting that excessive amounts of the do** agent led to decreased activity, possibly due to an excess of HMT causing an abundance of vacancies that acted as recombination centers, thereby reducing the overall efficiency of the photocatalysis process. The co-doped catalyst displayed stability over four consecutive cycles, indicating its operational reliability for the photocatalytic reduction of CO2. Researchers employed DFT calculations to gain insight into the mechanism of intramolecular photo-induced electron transfer. The calculations focused on three representative fragments (F1, F2, F3) chosen due to their complex structures. Theoretical energy levels were determined based on the structural models of these fragments. The results revealed that fragments containing O atoms were electron donors, while fragments containing C atoms acted as electron acceptors. Among the fragments, the heterocycle with an O atom within its cyclic structure exhibited a stronger electron donor characteristic than the heterocycle with an O atom outside its cyclic structure. Notably, fragments F1 and F3 demonstrated a distinct spatial separation between the HOMO and the LUMO. In these fragments, the HOMO primarily localized on the heterocycle unit with an O atom, while the LUMO mainly resided on the heterocycle unit with an ethylene group. On the other hand, fragment F2 exhibited a more uniform distribution of the HOMO throughout the fragment, while the LUMO predominantly localized on the heterocycle units with the ethylene group, indicating a partial spatial separation between the HOMO and LUMO. The clear spatial separation of the HOMO and LUMO in fragments F1 and F3 facilitated efficient charge separation during the transition, making them highly suitable for photocatalytic reactions. Consequently, in the context of donor–acceptor approaches in g-C3N4, a higher proportion of F1 and F3 fragments contributed to the improved photocatalytic performance of the co-doped catalyst [227]. A successful design and manufacturing method was employed to address the limitations of g-C3N4 nanosheets, including poor visible light absorption and low efficiency in charge transportation. This involved co-do** the nanosheets with C and O, resulting in improved performance in photocatalytic applications, particularly in the production of H2 and the reduction of CO2. The photocatalytic system strongly preferred CO as the primary product during CO2 reduction, with the co-doped g-C3N4 achieving a remarkable CO selectivity of 55.2 μmolg−1. This enhanced performance was attributed to the efficient extraction and transfer of electrons facilitated by introducing C and O dopants, along with enhanced absorption of visible light. In the CO2 reduction experiments using a mixture of water, methyl cyanide (MeCN), and TEOA, all tested g-C3N4 photocatalysts detected the presence of CO, CH4, and H2. However, the system exhibited a strong inclination towards CO production, with the co-doped g-C3N4 achieving a CO formation rate of 55.2 μmolg−1 after 12 h of visible light exposure. This rate was 2.0 and 9.5 times higher than pure g-C3N4, respectively. The slower reaction kinetics in the subsequent steps after CO formation limited the occurrence of further reactions before CO desorbed from the surface of the tubular g-C3N4, resulting in CO being the dominant product. At the same time other compounds like CH3OH, CH3CH2OH, or HCOOH were not detected. The co-doped g-C3N4, with C and O as dopants, exhibited a CO formation rate of 4.6 μmolg−1 h−1 during CO2 reduction, displaying an impressive selectivity of 92.5% for CO, surpassing previous g-C3N4-based photocatalysts. These findings confirm that CO formation result from the photocatalytic reduction of CO2. Furthermore, the synthesized co-doped g-C3N4 photocatalysts demonstrated excellent stability over three consecutives testing cycles. Based on the obtained results, the mechanism of CO2 reduction was analyzed, revealing that the key processes involved the reduction reactions facilitated by protons. This process included the transfer of multiple electrons, where photoexcited electrons in the CB reduced CO2 to generate CO and CH4. However, due to the lower electron requirement, CO was preferentially formed over other products that involved more complex reaction pathways. Additionally, the holes in the VB oxidized TEOA to TEOA+, while the electrons in the CB reduced CO2 to CO and CH4, with CO being the primary product [346].
To address the challenges of low CO2 activation capability and limited concentration of charge carriers in g- g-C3N4, researchers developed a new type of g-C3N4 by introducing C vacancies through heat treatment in an NH3 atmosphere. This modified g-C3N4 with enriched C vacancies showed a CO2 to CO conversion rate more than twice that of pristine g-C3N4. The improved CO2 reduction performance of the modified g-C3N4 can be attributed to enhanced CO2 adsorption/activation, an elevated CB, and increased concentration and lifetime of charge carriers. Moreover, introducing C vacancies helped to mitigate the exciton effect and promote the generation of charge carriers. These findings provide insights into enhancing CO2 photoreduction in g-C3N4 through structural adjustments and offer possibilities for manipulating the exciton effect and charge carrier concentration to facilitate photocatalytic reactions involving electron–hole separation and charge carriers. During the reaction, both photocatalysts exclusively produced CO as the carbonaceous product. The CO evolution rate on the modified g-C3N4 was approximately 4.18 mmolg−1 h−1, nearly 3.3 times higher than that on pristine g-C3N4 (approximately 1.28 mmol g−1 h−1) within the first hour (Fig. 13f). However, the increase was not as significant under full spectrum light. A comparative analysis examined the CO2 adsorption/activation, light-harvesting, and charge separation and transfer capabilities of pristine g-C3N4 and the modified g-C3N4 with C vacancies. Pristine g-C3N4 displayed inadequate active sites for CO2 adsorption/activation, as indicated by the CO2 temperature programmed desorption (CO2-TPD) results revealed a single desorption peak at around 80 °C. In contrast, the modified g-C3N4 exhibited two desorption peaks at approximately -85 °C and—330 °C. The lower temperature peak indicated physical adsorption of CO2, with a slightly higher desorption temperature than pristine g-C3N4. The peak at 330 °C was attributed to the chemisorption of CO2, likely facilitated by the increased basic sites resulting from more N–H groups in the modified g-C3N4 with C vacancies. DFT calculations supported these findings, showing higher CO2 adsorption energies on the modified g-C3N4 with C vacancies compared to pristine g-C3N4. Overall, the presence of C vacancies in g-C3N4 enhances CO2 adsorption/activation and improves the CO2 to CO conversion rate, underscoring the significance of C vacancies in facilitating CO2 photocatalytic reduction [230]. The performance of g-C3N4-based photocatalytic systems can be significantly improved by controlling their morphology and adjusting surface vacancies. This study aimed to understand the impact of N vacancies in the atomic layers of g-C3N4 on the photoreduction of CO2. By introducing an NH4Cl gas template, which facilitated efficient charge transportation, the researchers examined the influence of N vacancies on the photocatalytic properties. Incorporating the gas template disrupted the interactions between adjacent g-C3N4 layers, resulting in the desired structure. Additionally, N vacancies in the ultrathin nanosheets had notable effects on their optoelectronic characteristics. The researchers made several key findings regarding the N vacancies: First, a higher density of N vacancies enhanced the ability of g-C3N4 to reduce CO2. Second, the N vacancies allowed for extended absorption of long-wavelength visible light due to the strategic positioning of a midgap state below the CB. Third, the midgap state acted as an effective electron reservoir, prolonging the radiative recombination of electron–hole pairs. Consequently, g-C3N4 with N defects exhibited significant improvements in photocatalytic performance. The yield of CH4 evolution was 3.16 times higher compared to pristine ultrathin g-C3N4 and 5.14 times higher compared to bulk g-C3N4. The broader absorption spectra and longer lifetime of charge carriers demonstrated the crucial role of N defect states in enhancing the photocatalytic activity of g-C3N4. The CB edge of both ultrathin g-C3N4 and N defect-modified materials favored the production of CH4 through CO2 reduction, while H2O was oxidized to O2. The rate of CH4 evolution increased with a higher density of N vacancies, resulting in a CH4 evolution rate of 9.26 μmolg−1, which was 3.16 times higher than that of ultrathin g-C3N4. Photostability tests showed a relatively consistent CH4 evolution yield throughout the cycles, indicating the resistance of N defect sites to radical species and their ability to sustain prolonged light exposure. Based on the results, the researchers proposed a mechanistic understanding of how N vacancies contribute to improved photoactivity. N vacancies created a midgap state closer to the CB edge, characterized by a band tail energy between 1.2 and 1.3 eV. This unique positioning allowed the catalyst to effectively capture both short- and long-wavelength photons within the visible light range, leading to a two-step electron excitation process. The midgap state acted as an electron reservoir, efficiently trap** unstable electrons and preventing their direct recombination with holes. When sufficient excitation energy was available, the trapped electrons were excited from the midgap state to the CB, actively participating in the CO2 reduction process [237]. A simple one-step method was utilized to prepare 3D macroporous g-C3N4 with C vacancies, using polymethylmethacrylate as a template. This approach effectively increased the available reaction sites on the surface of g-C3N4 and expanded its absorption range for visible light. The modified g-C3N4 demonstrated remarkable improvements in reducing CO2 compared to the unmodified counterpart. The unique pore structure of the material played a significant role in enhancing light utilization efficiency and providing a greater number of active surface sites for catalysis. The performance of the modified samples was evaluated based on their capacity to reduce CO2, with the primary product being CH3OH. Notably, the modified catalyst exhibited a CH3OH generation rate of 7.5 μmolh−1 g−1, which surpassed the rate achieved by the unmodified g-C3N4 fourfold. This outcome underscores the importance of the macroporous structure, high specific surface area, and presence of C vacancies in facilitating the capture and photoreduction of CO2 molecules on the catalyst's surface. The efficiency of the photoreduction process is closely linked to the material’s electronic structure. Analysis of the electron configuration revealed that the CBM of the modified g-C3N4 was shifted to -2.1 eV, in contrast to the CBM value of -1.6 eV for bulk g-C3N4. This considerable shift in CBM creates a strong driving force for the conversion of CO2 by raising the reduction potential of the photogenerated electrons. The C vacancies act as effective electron traps, promoting photogenerated electrons localization and enhancing surface carriers utilization. These combined mechanisms synergistically contribute to the improved catalytic activity of the modified g-C3N4 in the photoreduction of CO2 [347].
6 Conclusion and future prospectives
g-C3N4 has attracted considerable attention as a photocatalyst in areas like photocatalytic water splitting and CO2 reduction. It possesses desirable properties, such as responsiveness to visible light, suitable band gap, excellent redox ability, metal-free nature, environmental friendliness, chemical and thermal stability, ease of fabrication, and modifiability. However, pristine g-C3N4 has limitations that hinder its photocatalytic efficiency, including insufficient sunlight absorption, low surface area, and fast recombination of electron–hole pairs. To overcome these limitations, researchers have explored nonmetal do** as a promising approach to enhance the photocatalytic activity of g-C3N4. This review summarizes recent advancements in develo** efficient and cost-effective systems based on doped g-C3N4. Nonmetal do**, co-do**, and vacancy engineering have been investigated to improve photocatalytic performance by enhancing light absorption, facilitating charge separation and transport, and extending the lifetime of charge carriers. By introducing external dopant atoms into g-C3N4, the electronic structure and energy levels can be adjusted, resulting in increased light responsiveness and improved charge separation. Nonmetal do** creates new energy levels within the band gap, broadening the spectral response and reducing the recombination rate of electron- hole pairs. However, these new energy bands can also act as recombination centers, decreasing quantum efficiencies. Nonetheless, nonmetal do** effectively modulates light absorption, redox potentials, and the mobility of photo-induced charge carriers. Co-do** offers additional benefits by combining the positive effects of individual dopants, thereby enhancing the structural and optical properties of g-C3N4 and ultimately improving its photocatalytic performance. Despite promising results, the development of nonmetal-doped g-C3N4 in photocatalysis is still in its early stages, and some challenges need to be addressed. The review also highlights significant advancements in vacancy creation in g-C3N4, which have greatly improved photocatalytic energy performance. The introduction of vacancies or dopants effectively modifies the material’s surface, optical, and electrical properties, resulting in enhanced photocatalytic performance in water splitting and CO2 reduction. Creating vacancies in g-C3N4 nanomaterials brings about significant changes, such as tunable bandgaps, defect-induced midgaps, increased surface area, suppressed recombination of electron–hole pairs, and improved adsorption and activation of reactant molecules. Do** with elements like B, O, S, and P and engineering N and C vacancies has increased reactant molecules’ affinity and binding energy, thereby enhancing uptake. These modifications enable the selective production of desired products in solar energy conversion.
However, despite extensive research and a positive outlook, the field of doped g-C3N4 still faces numerous scientific and engineering challenges that must be overcome. Several notable problems and future suggestions for the advancement of the do** effect in g-C3N4 include:
-
1.
Do** modifications of g-C3N4 can expand its ability to absorb visible light but often result in an indirect band gap, leading to insufficient light absorption intensity in the visible region. It is crucial to develop dopants and methods that create a direct band gap to enhance the absorption properties of g-C3N4. By introducing dopants into g-C3N4 materials, coordination unsaturated centers can be formed, providing highly active sites for the activation and chemisorption of reactant molecules. Future research should focus on specific techniques that anchor structural units to create single-atom catalysts with improved reaction kinetics. Ensuring the formation of uniform do** centers and preventing their disappearance or transformation are essential factors in this approach.
-
2.
The exploration of incorporating non-metal do** with the production of nanostructures, such as nanosheets, mesostructured, nanorods, nanotubes, and nanofibers, opens up a new and exciting direction. Nanostructured forms of g-C3N4, especially nanosheets, offer several advantages over bulk g-C3N4, including a larger specific surface area, improved transfer of electron–hole pairs, and enhanced efficiency in separating carriers. Template methods, like hard and soft templates, can synthesize mesoporous g-C3N4, increasing specific surface area and electron capture sites. However, exploring alternative techniques that minimize using environmentally harmful strong acids or alkalis is necessary. Choosing an appropriate do** method should align with the structural characteristics of g-C3N4. Interlayer do**, which uses a layered structure similar to graphite, can enhance electron transfer capabilities. On the other hand, in-plane do** can modify the energy barriers for surface reactions, allowing for specific adjustments by targeting different do** sites.
-
3.
Further exploration is needed to understand the relationship between dopants and the various morphologies of g-C3N4 in terms of their impact on photocatalytic activity. This requires conducting experimental and computational analyses to investigate dopant modification, catalytic performance, active site properties, and underlying reaction mechanisms. Using computational chemistry can provide valuable insights into how do** influences the electronic structure, adsorption energy, and Gibbs free energy changes during photocatalysis. One promising avenue for improving the photocatalytic efficiency of nonmetal-doped g-C3N4 involves studying the modulation of the orientations of the HOMO and LUMO to encompass oxidation and reduction potentials while simultaneously reducing the band gap and enhancing the separation of photo-generated electrons and holes. To advance this field, it is crucial to understand the origins of visible light absorption caused by element do**, the resulting chemical states, and the specific locations of dopants.
-
4.
The current methods used to characterize materials have limitations when accurately identifying and quantifying do** in g-C3N4. Although advanced techniques like EPR, XPS, SFG, HRTEM, AFM, and STEM have been helpful, there is a need for more precise and refined characterization methods to understand the different types of dopants present in g-C3N4 fully. It is crucial to establish reliable techniques or systematic approaches to distinguish between dopant types, concentrations, and locations to deepen our understanding of their specific catalytic activities. Another unexplored aspect of do** materials relates to the structural changes that vacancies undergo during catalytic processes. Vacancies, known for their high energy and reactivity, can experience dynamic structural transformations during reactions. Investigating the stability of dopants throughout these processes contributes significantly to our understanding of their catalytic activity. Therefore, it is essential to increase the use of in situ/operando analyses (such as FTIR, Raman, XAFS, XPS, and SAS) to shed light on the dynamic structure of doped g-C3N4.
-
5.
The innovative concept of merging light-driven catalysis with electrochemical or bio-catalytic reactions offers promising opportunities. Biological systems utilize enzymes and reductive pathways to produce long-chain hydrocarbons, and by integrating biocatalysts with light-driven catalysis, we can capitalize on the unique benefits of each approach. The versatile properties of doped g-C3N4, including its customizable structure, tunable surface properties, and flexible nature, make it an ideal candidate for synergistic interactions with cellular organisms. This integration opens up possibilities for multifunctional photo redox applications, maximizing their potential.
Here, we have emphasized a crucial facet of sustainable energy research, focusing on the pivotal role of surface chemistry in enhancing the performance of g-C3N4 as a metal-free semiconductor photocatalyst for clean energy conversion. Our manuscript delves into recent breakthroughs in the precise engineering of single g-C3N4 at the atomic level, with a pronounced focus on enhancing its surface chemistry through various do** techniques. These techniques encompass nonmetal and bi-nonmetal do** and the strategic creation of vacancies within the polymer framework. By finely tuning the surface chemical properties, we aim to address issues related to charge carrier dynamics, specific surface area, and light absorption properties. To provide a rigorous and comprehensive analysis, computational simulations based on first principles and DFT were employed to investigate the influence of do** and vacancy creation on the distribution of surface states, adsorption energies, and the interaction of g-C3N4 with target molecules. We highlight how surface chemistry modifications impact the efficiency of photocatalytic reactions, including water splitting and CO2 reduction, with a particular focus on product selectivity.
Collectively adhering to these guidelines can extend valuable insights for designing efficient and continuous catalytic systems in artificial photosynthesis. With collaborative efforts spanning various disciplines such as materials engineering, chemical engineering, electrical engineering, computational science, chemistry, and physics and involvement from industrial stakeholders, we hold strong confidence that this approach will establish a groundbreaking paradigm. It will propel scalable doped g-C3N4 to the forefront and usher in a new era of renewable energy in the coming years. Furthermore, the future integration of machine learning, big data, and artificial intelligence can enhance the do** engineering of g-C3N4 for light-driven energy catalysis. Through comprehensive collection and analysis of experimental and simulation results, we firmly believe that do** engineering in g-C3N4 photocatalysts will rectify imperfections observed in their pristine counterparts. This, in turn, will unlock unprecedented opportunities for solar energy conversion and transform visionary concepts into tangible breakthroughs. Ultimately, it will revolutionize the field of photocatalysis with remarkable advancements in specialized applications.
Abbreviations
- SDG7:
-
Sustainable Development Goal
- g-C3N4 :
-
Graphitic carbon nitride
- CB:
-
Conduction band
- DOS:
-
Density of states
- DFT:
-
Density functional theory
- H2 :
-
Hydrogen
- NPs:
-
Nanoparticles
- EA:
-
Elemental analysis
- MAS NMR:
-
Magic angle spinning nuclear magnetic resonance
- LDA:
-
Local density approximation
- GGA:
-
Generalized gradient approximation
- XRD:
-
X-ray powder diffraction
- N:
-
Nitrogen
- XPS:
-
X-ray photoelectron spectroscopy
- TON:
-
Turnover number
- LUMO:
-
Lowest unoccupied molecular orbital
- HOMO:
-
Highest occupied molecular orbital
- B:
-
Boron
- CH3CH2OH:
-
Ethanol
- O2 :
-
Oxygen
- XANES:
-
X-ray absorption near-edge structure
- C:
-
Carbon
- VB:
-
Valance band
- UVDRS:
-
Ultraviolet-visible diffuse reflectance spectroscopy
- O:
-
Oxygen
- TEM:
-
Transmission electron microscopy
- FDTD:
-
Finite-difference time-domain
- PVP:
-
Polyvinylpyrrolidone
- AFM:
-
Atomic force microscopy
- TRPL:
-
Time-resolved photoluminescence
- P:
-
Phosphorus
- 31P:
-
Phosphorus-31
- F:
-
Fluorine
- SEM:
-
Scanning electron microscope
- EIS:
-
Electrochemical impedance spectroscopy
- NaH2PO2·H2O:
-
Sodium hypophosphite monohydrate
- Cl:
-
Chlorine
- Br:
-
Bromine
- I:
-
Iodine
- CP:
-
Cross-polarization
- LSPR:
-
Localized surface plasmon resonance
- PEC:
-
Photoelectrochemical
- AQY:
-
Apparent quantum yield
- TEOA:
-
Triethanolamine
- CH3OH:
-
Methanol
- HCOOH:
-
Formic acid
- HCHO:
-
Formaldehyde
- CH4 :
-
Methane
- HMT:
-
Hexamethylenetetramine
- MeCN:
-
Methyl cyanide
- CO2-TPD:
-
CO2 temperature-programmed desorption
References
Falcone PM, Hiete M, Sapio A (2021) Hydrogen economy and sustainable development goals: review and policy insights. Curr Opin Green Sustain Chem 31:100506. https://doi.org/10.1016/j.cogsc.2021.100506
Maggio G, Nicita A, Squadrito G (2019) How the hydrogen production from RES could change energy and fuel markets: a review of recent literature. Int J Hydrogen Energy 44:11371–11384. https://doi.org/10.1016/j.ijhydene.2019.03.121
Penner SS (2006) Steps toward the hydrogen economy. Energy 31:33–43. https://doi.org/10.1016/j.energy.2004.04.060
Staffell I, Scamman D, Velazquez Abad A, Balcombe P, Dodds PE, Ekins P, Shah N, Ward KR (2019) The role of hydrogen and fuel cells in the global energy system. Energy Environ Sci 12:463–491. https://doi.org/10.1039/C8EE01157E
Wesseling JH, Lechtenböhmer S, Åhman M, Nilsson LJ, Worrell E, Coenen L (2017) The transition of energy intensive processing industries towards deep decarbonization: Characteristics and implications for future research. Renew Sust Energ Rev 79:1303–1313. https://doi.org/10.1016/j.rser.2017.05.156
von Stechow C, Minx JC, Riahi K, Jewell J, McCollum DL, Callaghan MW, Bertram C, Luderer G, Baiocchi G (2016) 2 °C and SDGs: united they stand, divided they fall? Environ Res Lett 11:034022. https://doi.org/10.1088/1748-9326/11/3/034022
Grubler A, Wilson C, Bento N, Boza-Kiss B, Krey V, McCollum DL, Rao ND, Riahi K, Rogelj J, De Stercke S, Cullen J, Frank S, Fricko O, Guo F, Gidden M, Havlík P, Huppmann D, Kiesewetter G, Rafaj P, Schoepp W, Valin H (2018) A low energy demand scenario for meeting the 1.5 °C target and sustainable development goals without negative emission technologies. Nat Energy 3:515–527. https://doi.org/10.1038/s41560-018-0172-6
Dogan E, Madaleno M, Taskin D, Tzeremes P (2022) Investigating the spillovers and connectedness between green finance and renewable energy sources. Renew Energ 197:709–722. https://doi.org/10.1016/j.renene.2022.07.131
Olabi AG, Abdelkareem MA (2022) Renewable energy and climate change. Renew Sust Energ Rev 158:112111. https://doi.org/10.1016/j.rser.2022.112111
Osman AI, Chen L, Yang M, Msigwa G, Farghali M, Fawzy S, Rooney DW, Yap P-S (2023) Cost, environmental impact, and resilience of renewable energy under a changing climate: a review. Environ Chem Lett 21:741–764. https://doi.org/10.1007/s10311-022-01532-8
Yue M, Lambert H, Pahon E, Roche R, Jemei S, Hissel D (2021) Hydrogen energy systems: a critical review of technologies, applications, trends and challenges. Renew Sust Energ Rev 146:111180. https://doi.org/10.1016/j.rser.2021.111180
Lebrouhi BE, Djoupo JJ, Lamrani B, Benabdelaziz K, Kousksou T (2022) Global hydrogen development - A technological and geopolitical overview. Int J Hydrogen Energy 47:7016–7048. https://doi.org/10.1016/j.ijhydene.2021.12.076
Rasul MG, Hazrat MA, Sattar MA, Jahirul MI, Shearer MJ (2022) The future of hydrogen: Challenges on production, storage and applications. Energy Convers Manage 272:116326. https://doi.org/10.1016/j.enconman.2022.116326
Egeland-Eriksen T, Hajizadeh A, Sartori S (2021) Hydrogen-based systems for integration of renewable energy in power systems: achievements and perspectives. Int J Hydrogen Energy 46:31963–31983. https://doi.org/10.1016/j.ijhydene.2021.06.218
**kuo L, Xue H (2022) Comparative analysis on similarities and differences of hydrogen energy development in the World’s top 4 largest economies: a novel framework. Int J Hydrogen Energy 47:9485–9503. https://doi.org/10.1016/j.ijhydene.2022.01.038
Hren R, Vujanović A, Van Fan Y, Klemeš JJ, Krajnc D, Čuček L (2023) Hydrogen production, storage and transport for renewable energy and chemicals: an environmental footprint assessment. Renew Sust Energ Rev 173:113113. https://doi.org/10.1016/j.rser.2022.113113
Gordon JA, Balta-Ozkan N, Nabavi SA (2023) Socio-technical barriers to domestic hydrogen futures: repurposing pipelines, policies, and public perceptions. Appl Energy 336:120850. https://doi.org/10.1016/j.apenergy.2023.120850
Gong J, Li C, Wasielewski MR (2019) Advances in solar energy conversion. Chem Soc Rev 48:1862–1864. https://doi.org/10.1039/C9CS90020A
Zhang Y, Ren J, Pu Y, Wang P (2020) Solar energy potential assessment: a framework to integrate geographic, technological, and economic indices for a potential analysis. Renew Energ 149:577–586. https://doi.org/10.1016/j.renene.2019.12.071
Lewis NS (2016) Research opportunities to advance solar energy utilization. Science 351:1920. https://doi.org/10.1126/science.aad1920
Lewis NS (2015) Introduction: solar energy conversion. Chem Rev 115:12631–12632. https://doi.org/10.1021/acs.chemrev.5b00654
Nelson N, Ben-Shem A (2004) The complex architecture of oxygenic photosynthesis. Nat Rev Mol Cell Biol 5:971–982. https://doi.org/10.1038/nrm1525
El-Khouly ME, El-Mohsnawy E, Fukuzumi S (2017) Solar energy conversion: From natural to artificial photosynthesis. J Photochem Photobiol C 31:36–83. https://doi.org/10.1016/j.jphotochemrev.2017.02.001
Lv J, **e J, Mohamed AGA, Zhang X, Wang Y (2022) Photoelectrochemical energy storage materials: design principles and functional devices towards direct solar to electrochemical energy storage. Chem Soc Rev 51:1511–1528. https://doi.org/10.1039/D1CS00859E
Khan SN, Yang Z, Dong W, Zhao M (2022) Cost and technology readiness level assessment of emerging technologies, new perspectives, and future research directions in H2 production. Sustain Energy Fuels 6:4357–4374. https://doi.org/10.1039/D2SE00988A
Dolle C, Neha N, Coutanceau C (2022) Electrochemical hydrogen production from biomass. Curr Opin Electrochem 31:100841. https://doi.org/10.1016/j.coelec.2021.100841
Groenemans H, Saur G, Mittelsteadt C, Lattimer J, Xu H (2022) Techno-economic analysis of offshore wind PEM water electrolysis for H2 production. Curr Opin Chem Eng 37:100828. https://doi.org/10.1016/j.coche.2022.100828
Song H, Luo S, Huang H, Deng B, Ye J (2022) Solar-driven hydrogen production: recent advances, challenges, and future perspectives. ACS Energy Lett 7:1043–1065. https://doi.org/10.1021/acsenergylett.1c02591
Ishaq H, Dincer I, Crawford C (2022) A review on hydrogen production and utilization: challenges and opportunities. Int J Hydrogen Energy 47:26238–26264. https://doi.org/10.1016/j.ijhydene.2021.11.149
Qureshi F, Yusuf M, Kamyab H, Vo D-VN, Chelliapan S, Joo S-W, Vasseghian Y (2022) Latest eco-friendly avenues on hydrogen production towards a circular bioeconomy: currents challenges, innovative insights, and future perspectives. Renew Sust Energ Rev 168:112916. https://doi.org/10.1016/j.rser.2022.112916
White JL, Baruch MF, Pander JE III, Hu Y, Fortmeyer IC, Park JE, Zhang T, Liao K, Gu J, Yan Y, Shaw TW, Abelev E, Bocarsly AB (2015) Light-driven heterogeneous reduction of carbon dioxide: photocatalysts and photoelectrodes. Chem Rev 115:12888–12935. https://doi.org/10.1021/acs.chemrev.5b00370
Etacheri V, Di Valentin C, Schneider J, Bahnemann D, Pillai SC (2015) Visible-light activation of TiO2 photocatalysts: advances in theory and experiments. J Photochem Photobiol C 25:1–29. https://doi.org/10.1016/j.jphotochemrev.2015.08.003
She X, Wu J, Xu H, Zhong J, Wang Y, Song Y, Nie K, Liu Y, Yang Y, Rodrigues M-TF, Vajtai R, Lou J, Du D, Li H, Ajayan PM (2017) High efficiency photocatalytic water splitting using 2D α-Fe2O3/g-C3N4 Z-scheme catalysts. Adv Energy Mater 7:1700025. https://doi.org/10.1002/aenm.201700025
Ning X, Lu G (2020) Photocorrosion inhibition of CdS-based catalysts for photocatalytic overall water splitting. Nanoscale 12:1213–1223. https://doi.org/10.1039/C9NR09183A
Chen T, Liu L, Hu C, Huang H (2021) Recent advances on Bi2WO6-based photocatalysts for environmental and energy applications. Chinese J Catal 42:1413–1438. https://doi.org/10.1016/S1872-2067(20)63769-X
Babu P, Mohanty S, Naik B, Parida K (2019) Serendipitous assembly of mixed phase BiVO4 on B-doped g-C3N4: An appropriate p–n heterojunction for photocatalytic O2 evolution and Cr(VI) reduction. Inorg Chem 58:12480–12491. https://doi.org/10.1021/acs.inorgchem.9b02309
Noda Y, Lee B, Domen K, Kondo JN (2008) Synthesis of crystallized mesoporous tantalum oxide and its photocatalytic activity for overall water splitting under ultraviolet light irradiation. Chem Mater 20:5361–5367. https://doi.org/10.1021/cm703202n
Fu J, Wang F, **ao Y, Yao Y, Feng C, Chang L, Jiang C-M, Kunzelmann VF, Wang ZM, Govorov AO, Sharp ID, Li Y (2020) Identifying performance-limiting deep traps in Ta3N5 for solar water splitting. ACS Cat 10:10316–10324. https://doi.org/10.1021/acscatal.0c02648
**ao J, Nishimae S, Vequizo JJM, Nakabayashi M, Hisatomi T, Li H, Lin L, Shibata N, Yamakata A, Inoue Y, Domen K (2022) Enhanced overall water splitting by a zirconium-doped TaON-based photocatalyst. Angew Chem Int Ed 61:e202116573. https://doi.org/10.1002/anie.202116573
Wang X, Blechert S, Antonietti M (2012) Polymeric graphitic carbon nitride for heterogeneous photocatalysis. ACS Cat 2:1596–1606. https://doi.org/10.1021/cs300240x
Ong W-J, Tan L-L, Ng YH, Yong S-T, Chai S-P (2016) Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem Rev 116:7159–7329. https://doi.org/10.1021/acs.chemrev.6b00075
Dong G, Zhang Y, Pan Q, Qiu J (2014) A fantastic graphitic carbon nitride (g-C3N4) material: electronic structure, photocatalytic and photoelectronic properties. J Photochem Photobiol C 20:33–50. https://doi.org/10.1016/j.jphotochemrev.2014.04.002
Cao S, Low J, Yu J, Jaroniec M (2015) Polymeric photocatalysts based on graphitic carbon nitride. Adv Mater 27:2150–2176. https://doi.org/10.1002/adma.201500033
Liu J, Wang H, Antonietti M (2016) Graphitic carbon nitride “reloaded”: emerging applications beyond (photo)catalysis. Chem Soc Rev 45:2308–2326. https://doi.org/10.1039/C5CS00767D
Wang Y, Liu L, Ma T, Zhang Y, Huang H (2021) 2D graphitic carbon nitride for energy conversion and storage. Adv Funct Mater 31:2102540. https://doi.org/10.1002/adfm.202102540
Wang X, Maeda K, Thomas A, Takanabe K, **n G, Carlsson JM, Domen K, Antonietti M (2009) A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat Mater 8:76–80. https://doi.org/10.1038/nmat2317
Zhu B, Zhang L, Cheng B, Yu J (2018) First-principle calculation study of tri-s-triazine-based g-C3N4: a review. Appl Catal B 224:983–999. https://doi.org/10.1016/j.apcatb.2017.11.025
Brito WH, da Silva-Araújo J, Chacham H (2015) g-C3N4 and others: Predicting new nanoporous carbon nitride planar structures with distinct electronic properties. J Phys Chem C 119:19743–19751. https://doi.org/10.1021/acs.jpcc.5b02543
Majdoub M, Anfar Z, Amedlous A (2020) Emerging chemical functionalization of g-C3N4: covalent/noncovalent modifications and applications. ACS Nano 14:12390–12469. https://doi.org/10.1021/acsnano.0c06116
Wen J, **e J, Chen X, Li X (2017) A review on g-C3N4-based photocatalysts. Appl Surf Sci 391:72–123. https://doi.org/10.1016/j.apsusc.2016.07.030
Zheng Y, Liu J, Liang J, Jaroniec M, Qiao SZ (2012) Graphitic carbon nitride materials: controllable synthesis and applications in fuel cells and photocatalysis. Energy Environ Sci 5:6717–6731. https://doi.org/10.1039/C2EE03479D
Ling GZS, Ng S-F, Ong W-J (2022) Tailor-engineered 2D cocatalysts: Harnessing electron–hole redox center of 2D g-C3N4 photocatalysts toward solar-to-chemical conversion and environmental purification. Adv Funct Mater 32:2111875. https://doi.org/10.1002/adfm.202111875
Tian N, Huang H, Du X, Dong F, Zhang Y (2019) Rational nanostructure design of graphitic carbon nitride for photocatalytic applications. J Mater Chem A 7:11584–11612. https://doi.org/10.1039/C9TA01819K
Prasad C, Tang H, Liu Q, Bahadur I, Karlapudi S, Jiang Y (2020) A latest overview on photocatalytic application of g-C3N4 based nanostructured materials for hydrogen production. Int J Hydrogen Energy 45:337–379. https://doi.org/10.1016/j.ijhydene.2019.07.070
Li Y, Zhou M, Cheng B, Shao Y (2020) Recent advances in g-C3N4-based heterojunction photocatalysts. J Mater Sci Technol 56:1–17. https://doi.org/10.1016/j.jmst.2020.04.028
Li Y, **a Z, Yang Q, Wang L, **ng Y (2022) Review on g-C3N4-based S-scheme heterojunction photocatalysts. J Mater Sci Technol 125:128–144. https://doi.org/10.1016/j.jmst.2022.02.035
Zhang S, Gu P, Ma R, Luo C, Wen T, Zhao G, Cheng W, Wang X (2019) Recent developments in fabrication and structure regulation of visible-light-driven g-C3N4-based photocatalysts towards water purification: a critical review. Catal Today 335:65–77. https://doi.org/10.1016/j.cattod.2018.09.013
Podjaski F, Lotsch BV (2021) Optoelectronics meets optoionics: light storing carbon nitrides and beyond. Adv Energy Mater 11:2003049. https://doi.org/10.1002/aenm.202003049
Lu Q, Eid K, Li W, Abdullah AM, Xu G, Varma RS (2021) Engineering graphitic carbon nitride (g-C3N4) for catalytic reduction of CO2 to fuels and chemicals: strategy and mechanism. Green Chem 23:5394–5428. https://doi.org/10.1039/D1GC01303C
Dozzi MV, Selli E (2013) Do** TiO2 with p-block elements: effects on photocatalytic activity. J Photochem Photobiol C 14:13–28. https://doi.org/10.1016/j.jphotochemrev.2012.09.002
Putri LK, Ong W-J, Chang WS, Chai S-P (2015) Heteroatom doped graphene in photocatalysis: a review. Appl Surf Sci 358:2–14. https://doi.org/10.1016/j.apsusc.2015.08.177
Marschall R, Wang L (2014) Non-metal do** of transition metal oxides for visible-light photocatalysis. Catal Today 225:111–135. https://doi.org/10.1016/j.cattod.2013.10.088
Zhang S, Li J, Zeng M, Li J, Xu J, Wang X (2014) Bandgap engineering and mechanism study of nonmetal and metal ion codoped carbon nitride: C+Fe as an example. Chem Eur J 20:9805–9812. https://doi.org/10.1002/chem.201400060
Wu S, Yu H, Chen S, Quan X (2020) Enhanced photocatalytic H2O2 production over carbon nitride by do** and defect engineering. ACS Cat 10:14380–14389. https://doi.org/10.1021/acscatal.0c03359
Zhang J, Zhang M, Lin S, Fu X, Wang X (2014) Molecular do** of carbon nitride photocatalysts with tunable bandgap and enhanced activity. J Catal 310:24–30. https://doi.org/10.1016/j.jcat.2013.01.008
Li C-F, Ji Y-F, Wei Q-Y, Liu Z-B, Wu Y-Y, Chen LS, Han D-X, Niu L, Tao C-L, Qin D-D (2023) Defect and electronic structure engineering graphitic carbon nitride with dual-gas-phase reaction for visible-light-driven hydrogen evolution. ACS Appl Energy Mater 6:997–1007. https://doi.org/10.1021/acsaem.2c03448
Makaremi M, Grixti S, Butler KT, Ozin GA, Singh CV (2018) Band engineering of carbon nitride monolayers by N-type, P-type, and isoelectronic do** for photocatalytic applications. ACS Appl Mater Interfaces 10:11143–11151. https://doi.org/10.1021/acsami.8b01729
Chen P-W, Li K, Yu Y-X, Zhang W-D (2017) Cobalt-doped graphitic carbon nitride photocatalysts with high activity for hydrogen evolution. Appl Surf Sci 392:608–615. https://doi.org/10.1016/j.apsusc.2016.09.086
Wu C, Zhang X, **a Z, Shu M, Li H, Xu X, Si R, Rykov AI, Wang J, Yu S, Wang S, Sun G (2019) Insight into the role of Ni–Fe dual sites in the oxygen evolution reaction based on atomically metal-doped polymeric carbon nitride. J Mater Chem A 7:14001–14010. https://doi.org/10.1039/C9TA03163D
Ding G, Wang W, Jiang T, Han B, Fan H, Yang G (2013) Highly selective synthesis of phenol from benzene over a vanadium-doped graphitic carbon nitride catalyst. ChemCatChem 5:192–200. https://doi.org/10.1002/cctc.201200502
Yan Y, Yang Q, Shang Q, Ai J, Yang X, Wang D, Liao G (2022) Ru doped graphitic carbon nitride mediated peroxymonosulfate activation for diclofenac degradation via singlet oxygen. Chem Eng J 430:133174. https://doi.org/10.1016/j.cej.2021.133174
Meek TL, Garner LD (2005) Electronegativity and the bond triangle. J Chem Educ 82:325. https://doi.org/10.1021/ed082p325
Tong Y, Wei C, Li Y, Zhang Y, Lin W (2020) Unraveling the mechanisms of S-doped carbon nitride for photocatalytic oxygen reduction to H2O2. Phys Chem Chem Phys 22:21099–21107. https://doi.org/10.1039/D0CP03533E
Lin T-J, Chiu C-c (2020) Influence of nonmetal dopants on charge separation of graphitic carbon nitride by time-dependent density functional theory. Phys Chem Chem Phys 22:647–657. https://doi.org/10.1039/C9CP06175D
Wang Y, Wang H, Chen F, Cao F, Zhao X, Meng S, Cui Y (2017) Facile synthesis of oxygen doped carbon nitride hollow microsphere for photocatalysis. Appl Catal B 206:417–425. https://doi.org/10.1016/j.apcatb.2017.01.041
Li F, Han M, ** Y, Zhang L, Li T, Gao Y, Hu C (2019) Internal electric field construction on dual oxygen group-doped carbon nitride for enhanced photodegradation of pollutants under visible light irradiation. Appl Catal B 256:117705. https://doi.org/10.1016/j.apcatb.2019.05.007
Zhu Y, Chen Z, Gao Y, Hu C (2020) General synthesis of carbon and oxygen dual-doped graphitic carbon nitride via copolymerization for non-photochemical oxidation of organic pollutant. J Hazard Mater 394:122578. https://doi.org/10.1016/j.jhazmat.2020.122578
Li H, Liang Z, Deng Q, Hou W (2020) Band structure engineering of polymeric carbon nitride with oxygen/carbon codo** for efficient charge separation and photocatalytic performance. J Colloid Interface Sci 564:333–343. https://doi.org/10.1016/j.jcis.2019.12.131
Wei R, Tang N, Jiang L, Yang J, Guo J, Yuan X, Liang J, Zhu Y, Wu Z, Li H (2022) Bimetallic nanoparticles meet polymeric carbon nitride: Fabrications, catalytic applications and perspectives. Coord Chem Rev 462:214500. https://doi.org/10.1016/j.ccr.2022.214500
Li Y, Kong T, Shen S (2019) Artificial photosynthesis with polymeric carbon nitride: when meeting metal nanoparticles, single atoms, and molecular complexes. Small 15:1900772. https://doi.org/10.1002/smll.201900772
Jiang L, Yang J, Zhou S, Yu H, Liang J, Chu W, Li H, Wang H, Wu Z, Yuan X (2021) Strategies to extend near-infrared light harvest of polymer carbon nitride photocatalysts. Coord Chem Rev 439:213947. https://doi.org/10.1016/j.ccr.2021.213947
Malik R, Tomer VK (2021) State-of-the-art review of morphological advancements in graphitic carbon nitride (g-CN) for sustainable hydrogen production. Renew Sust Energ Rev 135:110235. https://doi.org/10.1016/j.rser.2020.110235
Wang S, Zhang J, Li B, Sun H, Wang S, Duan X (2022) Morphology-dependent photocatalysis of graphitic carbon nitride for sustainable remediation of aqueous pollutants: a mini review. J Environ Chem Eng 10:107438. https://doi.org/10.1016/j.jece.2022.107438
Zhu J, **ao P, Li H, Carabineiro SAC (2014) Graphitic carbon nitride: synthesis, properties, and applications in catalysis. ACS Appl Mater Interfaces 6:16449–16465. https://doi.org/10.1021/am502925j
Li Y, Li X, Zhang H, **ang Q (2020) Porous graphitic carbon nitride for solar photocatalytic applications. Nanoscale Horiz 5:765–786. https://doi.org/10.1039/D0NH00046A
Yang Z, Zhang Y, Schnepp Z (2015) Soft and hard templating of graphitic carbon nitride. J Mater Chem A 3:14081–14092. https://doi.org/10.1039/C5TA02156A
Huang D, Yan X, Yan M, Zeng G, Zhou C, Wan J, Cheng M, Xue W (2018) Graphitic carbon nitride-based heterojunction photoactive nanocomposites: applications and mechanism insight. ACS Appl Mater Interfaces 10:21035–21055. https://doi.org/10.1021/acsami.8b03620
Ren Y, Zeng D, Ong W-J (2019) Interfacial engineering of graphitic carbon nitride (g-C3N4)-based metal sulfide heterojunction photocatalysts for energy conversion: a review. Chinese J Catal 40:289–319. https://doi.org/10.1016/S1872-2067(19)63293-6
Sun W, Zhu J, Zheng Y (2021) Graphitic carbon nitride heterojunction photocatalysts for solar hydrogen production. Int J Hydrogen Energy 46:37242–37267. https://doi.org/10.1016/j.ijhydene.2021.09.019
Deng A, Sun Y, Gao Z, Yang S, Liu Y, He H, Zhang J, Liu S, Sun H, Wang S (2023) Internal electric field in carbon nitride-based heterojunctions for photocatalysis. Nano Energy 108:108228. https://doi.org/10.1016/j.nanoen.2023.108228
Kessler FK, Zheng Y, Schwarz D, Merschjann C, Schnick W, Wang X, Bojdys MJ (2017) Functional carbon nitride materials — design strategies for electrochemical devices. Nat Rev Mater 2:17030. https://doi.org/10.1038/natrevmats.2017.30
Mitchell E, Law A, Godin R (2021) Interfacial charge transfer in carbon nitride heterojunctions monitored by optical methods. J Photochem Photobiol C 49:100453. https://doi.org/10.1016/j.jphotochemrev.2021.100453
Wang S, Zhang J, Li B, Sun H, Wang S (2021) Engineered graphitic carbon nitride-based photocatalysts for visible-light-driven water splitting: a review. Energy Fuels 35:6504–6526. https://doi.org/10.1021/acs.energyfuels.1c00503
Miller TS, d’Aleo A, Suter T, Aliev AE, Sella A, McMillan PF (2017) Pharaoh’s serpents: new insights into a classic carbon nitride material. Z Anorg Allg Chem 643:1572–1580. https://doi.org/10.1002/zaac.201700268
Niu W, Yang Y (2018) Graphitic carbon nitride for electrochemical energy conversion and storage. ACS Energy Lett 3:2796–2815. https://doi.org/10.1021/acsenergylett.8b01594
Praus P (2022) A brief review of s-triazine graphitic carbon nitride. Carbon Lett 32:703–712. https://doi.org/10.1007/s42823-022-00319-9
Alwin E, Nowicki W, Wojcieszak R, Zieliński M, Pietrowski M (2020) Elucidating the structure of the graphitic carbon nitride nanomaterials via X-ray photoelectron spectroscopy and X-ray powder diffraction techniques. Dalton Trans 49:12805–12813. https://doi.org/10.1039/D0DT02325F
Liao G, Gong Y, Zhang L, Gao H, Yang G-J, Fang B (2019) Semiconductor polymeric graphitic carbon nitride photocatalysts: the “holy grail” for the photocatalytic hydrogen evolution reaction under visible light. Energy Environ Sci 12:2080–2147. https://doi.org/10.1039/C9EE00717B
Wang Y, Wang X, Antonietti M (2012) Polymeric graphitic carbon nitride as a heterogeneous organocatalyst: from photochemistry to multipurpose catalysis to sustainable chemistry. Angew Chem Int Ed 51:68–89. https://doi.org/10.1002/anie.201101182
Miller TS, Jorge AB, Suter TM, Sella A, Corà F, McMillan PF (2017) Carbon nitrides: synthesis and characterization of a new class of functional materials. Phys Chem Chem Phys 19:15613–15638. https://doi.org/10.1039/C7CP02711G
Franklin EC (1922) The ammono carbonic acids. J Am Chem Soc 44:486–509. https://doi.org/10.1021/ja01424a007
Chen D, Wang Y, Dronskowski R (2023) Computational design and theoretical properties of WC3N6, an H-free melaminate and potential multifunctional material. J Am Chem Soc 145:6986–6993. https://doi.org/10.1021/jacs.3c00631
Liu F, Tong Y, Li C, Liu X (2021) One-dimensional conjugated carbon nitrides: synthesis and structure determination by HRTEM and solid-state NMR. J Phys Chem Lett 12:10359–10365. https://doi.org/10.1021/acs.jpclett.1c02994
Görne AL, Scholz T, Kobertz D, Dronskowski R (2021) Deprotonating melamine to gain highly interconnected materials: Melaminate salts of potassium and rubidium. Inorg Chem 60:15069–15077. https://doi.org/10.1021/acs.inorgchem.1c02383
Zambon A, Mouesca JM, Gheorghiu C, Bayle PA, Pécaut J, Claeys-Bruno M, Gambarelli S, Dubois L (2016) s-Heptazine oligomers: promising structural models for graphitic carbon nitride. Chem Sci 7:945–950. https://doi.org/10.1039/C5SC02992A
Redemann CE, Lucas HJ (1940) Some derivatives of cyameluric acid and probable structures of melam, melem and melon. J Am Chem Soc 62:842–846. https://doi.org/10.1021/ja01861a038
Thomas A, Fischer A, Goettmann F, Antonietti M, Müller J-O, Schlögl R, Carlsson JM (2008) Graphitic carbon nitride materials: variation of structure and morphology and their use as metal-free catalysts. J Mater Chem 18:4893–4908. https://doi.org/10.1039/B800274F
Hao Q, Jia G, Wei W, Vinu A, Wang Y, Arandiyan H, Ni B-J (2020) Graphitic carbon nitride with different dimensionalities for energy and environmental applications. Nano Res 13:18–37. https://doi.org/10.1007/s12274-019-2589-z
Inagaki M, Tsumura T, Kinumoto T, Toyoda M (2019) Graphitic carbon nitrides (g-C3N4) with comparative discussion to carbon materials. Carbon 141:580–607. https://doi.org/10.1016/j.carbon.2018.09.082
Wood KN, O’Hayre R, Pylypenko S (2014) Recent progress on nitrogen/carbon structures designed for use in energy and sustainability applications. Energy Environ Sci 7:1212–1249. https://doi.org/10.1039/C3EE44078H
Komatsu T (2001) Prototype carbon nitrides similar to the symmetric triangular form of melon. J Mater Chem 11:802–803. https://doi.org/10.1039/B007165J
Komatsu T (2001) Attempted chemical synthesis of graphite-like carbon nitride. J Mater Chem 11:799–801. https://doi.org/10.1039/B007673M
Kroke E, Schwarz M, Horath-Bordon E, Kroll P, Noll B, Norman AD (2002) Tri-s-triazine derivatives. Part I. From trichloro-tri-s-triazine to graphitic C3N4 structures. New J Chem 26:508–512. https://doi.org/10.1039/B111062B
Schwarzer A, Saplinova T, Kroke E (2013) Tri-s-triazines (s-heptazines)—From a “mystery molecule” to industrially relevant carbon nitride materials. Coord Chem Rev 257:2032–2062. https://doi.org/10.1016/j.ccr.2012.12.006
Jürgens B, Irran E, Senker J, Kroll P, Müller H, Schnick W (2003) Melem (2,5,8-Triamino-tri-s-triazine), an important intermediate during condensation of melamine rings to graphitic carbon nitride: synthesis, structure determination by X-ray powder diffractometry, solid-state NMR, and theoretical studies. J Am Chem Soc 125:10288–10300. https://doi.org/10.1021/ja0357689
Sinha P, Boesch SE, Gu C, Wheeler RA, Wilson AK (2004) Harmonic vibrational frequencies: scaling factors for HF, B3LYP, and MP2 methods in combination with correlation consistent basis sets. J Phys Chem A 108:9213–9217. https://doi.org/10.1021/jp048233q
Stampfl C, Van de Walle CG (1999) Density-functional calculations for III-V nitrides using the local-density approximation and the generalized gradient approximation. Phys Rev B 59:5521–5535. https://doi.org/10.1103/PhysRevB.59.5521
Ye S, Wang R, Wu M-Z, Yuan Y-P (2015) A review on g-C3N4 for photocatalytic water splitting and CO2 reduction. Appl Surf Sci 358:15–27. https://doi.org/10.1016/j.apsusc.2015.08.173
Lau VW-H, Lotsch BV (2022) A tour-guide through carbon nitride-land: structure- and dimensionality-dependent properties for photo(electro)chemical energy conversion and storage. Adv Energy Mater 12:2101078. https://doi.org/10.1002/aenm.202101078
Lotsch BV, Schnick W (2007) New light on an old story: formation of melam during thermal condensation of melamine. Chem Eur J 13:4956–4968. https://doi.org/10.1002/chem.200601291
Lotsch BV, Schnick W (2004) Towards novel C-N materials: crystal structures of two polymorphs of guanidinium dicyanamide and their thermal conversion into melamine. New J Chem 28:1129–1136. https://doi.org/10.1039/B403788J
Huang C, Wen Y, Ma J, Dong D, Shen Y, Liu S, Ma H, Zhang Y (2021) Unraveling fundamental active units in carbon nitride for photocatalytic oxidation reactions. Nat Commun 12:320. https://doi.org/10.1038/s41467-020-20521-5
Algara-Siller G, Severin N, Chong SY, Björkman T, Palgrave RG, Laybourn A, Antonietti M, Khimyak YZ, Krasheninnikov AV, Rabe JP, Kaiser U, Cooper AI, Thomas A, Bojdys MJ (2014) Triazine-based graphitic carbon nitride: a two-dimensional semiconductor. Angew Chem Int Ed 53:7450–7455. https://doi.org/10.1002/anie.201402191
Lau VW-H, Mesch MB, Duppel V, Blum V, Senker J, Lotsch BV (2015) Low-molecular-weight carbon nitrides for solar hydrogen evolution. J Am Chem Soc 137:1064–1072. https://doi.org/10.1021/ja511802c
Gong Y, Li M, Li H, Wang Y (2015) Graphitic carbon nitride polymers: promising catalysts or catalyst supports for heterogeneous oxidation and hydrogenation. Green Chem 17:715–736. https://doi.org/10.1039/C4GC01847H
Chen Y, Wang X (2018) Template-free synthesis of hollow G-C3N4 polymer with vesicle structure for enhanced photocatalytic water splitting. J Phys Chem C 122:3786–3793. https://doi.org/10.1021/acs.jpcc.7b12496
Yan SC, Li ZS, Zou ZG (2009) Photodegradation performance of g-C3N4 fabricated by directly heating melamine. Langmuir 25:10397–10401. https://doi.org/10.1021/la900923z
Zhang G, Zhang J, Zhang M, Wang X (2012) Polycondensation of thiourea into carbon nitride semiconductors as visible light photocatalysts. J Mater Chem 22:8083–8091. https://doi.org/10.1039/C2JM00097K
Qin Z, Huang Z, Wang M, Liu D, Chen Y, Guo L (2020) Synergistic effect of quantum confinement and site-selective do** in polymeric carbon nitride towards overall water splitting. Appl Catal B 261:118211. https://doi.org/10.1016/j.apcatb.2019.118211
Lu P, Yang Y, Bai J, Zhao Z, Fu M, Hu X, He Y (2023) Defected mesoporous carbon nitride with quantum confinement effect for NO purification. J Alloys Compd 934:167825. https://doi.org/10.1016/j.jallcom.2022.167825
Zhou Z, Shen Y, Li Y, Liu A, Liu S, Zhang Y (2015) Chemical cleavage of layered carbon nitride with enhanced photoluminescent performances and photoconduction. ACS Nano 9:12480–12487. https://doi.org/10.1021/acsnano.5b05924
Lan Y, Li Z, Li D, Yan G, Yang Z, Guo S (2019) Graphitic carbon nitride synthesized at different temperatures for enhanced visible-light photodegradation of 2-naphthol. Appl Surf Sci 467–468:411–422. https://doi.org/10.1016/j.apsusc.2018.10.152
**ng J, Wang N, Li X, Wang J, Taiwaikuli M, Huang X, Wang T, Zhou L, Hao H (2022) Synthesis and modifications of g-C3N4-based materials and their applications in wastewater pollutants removal. J Environ Chem Eng 10:108782. https://doi.org/10.1016/j.jece.2022.108782
Jiang L, Yang J, Yuan X, Guo J, Liang J, Tang W, Chen Y, Li X, Wang H, Chu W (2021) Defect engineering in polymeric carbon nitride photocatalyst: Synthesis, properties and characterizations. Adv Colloid Interface Sci 296:102523. https://doi.org/10.1016/j.cis.2021.102523
Mills A, Le Hunte S (1997) An overview of semiconductor photocatalysis. J Photochem Photobiol A 108:1–35. https://doi.org/10.1016/S1010-6030(97)00118-4
Serpone N, Emeline AV (2012) Semiconductor photocatalysis — Past, present, and future Outlook. J Phys Chem Lett 3:673–677. https://doi.org/10.1021/jz300071j
Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Environmental applications of semiconductor photocatalysis. Chem Rev 95:69–96. https://doi.org/10.1021/cr00033a004
Kisch H (2013) Semiconductor photocatalysis—mechanistic and synthetic aspects. Angew Chem Int Ed 52:812–847. https://doi.org/10.1002/anie.201201200
Murray PRD, Cox JH, Chiappini ND, Roos CB, McLoughlin EA, Hejna BG, Nguyen ST, Ripberger HH, Ganley JM, Tsui E, Shin NY, Koronkiewicz B, Qiu G, Knowles RR (2022) Photochemical and electrochemical applications of proton-coupled electron transfer in organic synthesis. Chem Rev 122:2017–2291. https://doi.org/10.1021/acs.chemrev.1c00374
Prier CK, Rankic DA, MacMillan DWC (2013) Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem Rev 113:5322–5363. https://doi.org/10.1021/cr300503r
Hsu C-C, Wu NL (2005) Synthesis and photocatalytic activity of ZnO/ZnO2 composite. J Photochem Photobiol A 172:269–274. https://doi.org/10.1016/j.jphotochem.2004.12.014
Wang J, Wang Z, Huang B, Ma Y, Liu Y, Qin X, Zhang X, Dai Y (2012) Oxygen vacancy induced band-gap narrowing and enhanced visible light photocatalytic activity of ZnO. ACS Appl Mater Interfaces 4:4024–4030. https://doi.org/10.1021/am300835p
Schneider J, Matsuoka M, Takeuchi M, Zhang J, Horiuchi Y, Anpo M, Bahnemann DW (2014) Understanding TiO2 photocatalysis: mechanisms and materials. Chem Rev 114:9919–9986. https://doi.org/10.1021/cr5001892
Serpone N, Terzian R, Lawless D, Kennepohl P, Sauvé G (1993) On the usage of turnover numbers and quantum yields in heterogeneous photocatalysis. J Photochem Photobiol A 73:11–16. https://doi.org/10.1016/1010-6030(93)80027-7
Augugliaro V, Schiavello M, Palmisano L (1993) Rate of photon absorption and turnover number: two parameters for the comparison of heterogeneous photocatalytic systems in a quantitative way. Coord Chem Rev 125:173–181. https://doi.org/10.1016/0010-8545(93)85016-W
Hara M, Waraksa CC, Lean JT, Lewis BA, Mallouk TE (2000) Photocatalytic water oxidation in a buffered tris(2,2‘-bipyridyl)ruthenium complex-colloidal IrO2 system. J Phys Chem A 104:5275–5280. https://doi.org/10.1021/jp000321x
Hori H, Johnson FPA, Koike K, Ishitani O, Ibusuki T (1996) Efficient photocatalytic CO2 reduction using [Re(bpy) (CO)3{P(OEt)3}]+. J Photochem Photobiol A 96:171–174. https://doi.org/10.1016/1010-6030(95)04298-9
Gao C, Chen S, Wang Y, Wang J, Zheng X, Zhu J, Song L, Zhang W, **ong Y (2018) Heterogeneous single-atom catalyst for visible-light-driven high-turnover CO2 reduction: the role of electron transfer. Adv Mater 30:1704624. https://doi.org/10.1002/adma.201704624
Albini A, Fagnoni M (2008) 1908: Giacomo ciamician and the concept of green chemistry. Chemsuschem 1:63–66. https://doi.org/10.1002/cssc.200700015
Zhu S, Wang D (2017) Photocatalysis: basic principles, diverse forms of implementations and emerging scientific opportunities. Adv Energy Mater 7:1700841. https://doi.org/10.1002/aenm.201700841
Guo Q, Zhou C, Ma Z, Yang X (2019) Fundamentals of TiO2 photocatalysis: Concepts, mechanisms, and challenges. Adv Mater 31:1901997. https://doi.org/10.1002/adma.201901997
Pelaez M, Nolan NT, Pillai SC, Seery MK, Falaras P, Kontos AG, Dunlop PSM, Hamilton JWJ, Byrne JA, O’Shea K, Entezari MH, Dionysiou DD (2012) A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl Catal B 125:331–349. https://doi.org/10.1016/j.apcatb.2012.05.036
Bak T, Nowotny J, Rekas M, Sorrell CC (2002) Photo-electrochemical properties of the TiO2-Pt system in aqueous solutions. Int J Hydrogen Energy 27:19–26. https://doi.org/10.1016/S0360-3199(01)00090-8
Asahi R, Morikawa T, Irie H, Ohwaki T (2014) Nitrogen-doped titanium dioxide as visible-light-sensitive photocatalyst: designs, developments, and prospects. Chem Rev 114:9824–9852. https://doi.org/10.1021/cr5000738
Yamazaki Y, Takeda H, Ishitani O (2015) Photocatalytic reduction of CO2 using metal complexes. J Photochem Photobiol C 25:106–137. https://doi.org/10.1016/j.jphotochemrev.2015.09.001
Naik B, Kim SM, Jung CH, Moon SY, Kim SH, Park JY (2014) Enhanced H2 generation of Au-loaded, nitrogen-doped TiO2 hierarchical nanostructures under visible light. Adv Mater Interfaces 1:1300018. https://doi.org/10.1002/admi.201300018
Naik B, Moon SY, Kim SH, Park JY (2015) Enhanced photocatalytic generation of hydrogen by Pt-deposited nitrogen-doped TiO2 hierarchical nanostructures. Appl Surf Sci 354:347–352. https://doi.org/10.1016/j.apsusc.2015.05.102
Moon SY, Naik B, An K, Kim SM, Park JY (2016) Photocatalytic H2 generation on macro-mesoporous oxide-supported Pt nanoparticles. RSC Adv 6:18198–18203. https://doi.org/10.1039/C5RA25358F
Babu P, Naik B (2020) Cu–Ag bimetal alloy decorated SiO2@ TiO2 hybrid photocatalyst for enhanced H2 evolution and phenol oxidation under visible light. Inorg Chem 59:10824–10834. https://doi.org/10.1021/acs.inorgchem.0c01325
Moon SY, Naik B, Park JY (2016) Photocatalytic activity of metal-decorated SiO2@ TiO2 hybrid photocatalysts under water splitting. Korean J Chem Eng 33:2325–2329. https://doi.org/10.1007/s11814-016-0085-7
Cheng L, Zhang H, Li X, Fan J, **ang Q (2021) Carbon–graphitic carbon nitride hybrids for heterogeneous photocatalysis. Small 17:2005231. https://doi.org/10.1002/smll.202005231
Gong Y, Li M, Wang Y (2015) Carbon nitride in energy conversion and storage: recent advances and future prospects. Chemsuschem 8:931–946. https://doi.org/10.1002/cssc.201403287
Jia C, Yang L, Zhang Y, Zhang X, **ao K, Xu J, Liu J (2020) Graphitic carbon nitride films: emerging paradigm for versatile applications. ACS Appl Mater Interfaces 12:53571–53591. https://doi.org/10.1021/acsami.0c15159
**e K, Fang J, Li L, Deng J, Chen F (2022) Progress of graphite carbon nitride with different dimensions in the photocatalytic degradation of dyes: a review. J Alloys Compd 901:163589. https://doi.org/10.1016/j.jallcom.2021.163589
Mazzanti S, Savateev A (2020) Emerging concepts in carbon nitride organic photocatalysis. ChemPlusChem 85:2499–2517. https://doi.org/10.1002/cplu.202000606
Lin H, Wu J, Zhou F, Zhao X, Lu P, Sun G, Song Y, Li Y, Liu X, Dai H (2023) Graphitic carbon nitride-based photocatalysts in the applications of environmental catalysis. J Environ Sci (China) 124:570–590. https://doi.org/10.1016/j.jes.2021.11.017
Jourshabani M, Lee B-K, Shariatinia Z (2020) From traditional strategies to Z-scheme configuration in graphitic carbon nitride photocatalysts: recent progress and future challenges. Appl Catal B 276:119157. https://doi.org/10.1016/j.apcatb.2020.119157
Liang J, Jiang Z, Wong PK, Lee C-S (2021) Recent progress on carbon nitride and its hybrid photocatalysts for CO2 reduction. Solar RRL 5:2000478. https://doi.org/10.1002/solr.202000478
Hu C, Lin Y-R, Yang H-C (2019) Recent developments in graphitic carbon nitride based hydrogels as photocatalysts. Chemsuschem 12:1794–1806. https://doi.org/10.1002/cssc.201802257
Zhang L, Hou S, Wang T, Liu S, Gao X, Wang C, Wang G (2022) Recent advances in application of graphitic carbon nitride-based catalysts for photocatalytic nitrogen fixation. Small 18:2202252. https://doi.org/10.1002/smll.202202252
Pan H, Zhang Y-W, Shenoy VB, Gao H (2011) Effects of H-, N-, and (H, N)-do** on the photocatalytic activity of TiO2. J Phys Chem C 115:12224–12231. https://doi.org/10.1021/jp202385q
Yang Y, Li X-j, Chen J-t, Wang L-y (2004) Effect of do** mode on the photocatalytic activities of Mo/TiO2. J Photochem Photobiol A 163:517–522. https://doi.org/10.1016/j.jphotochem.2004.02.008
Peng S, Li Y, Jiang F, Lu G, Li S (2004) Effect of Be2+ do** TiO2 on its photocatalytic activity. Chem Phys Lett 398:235–239. https://doi.org/10.1016/j.cplett.2004.09.061
Aadil M, Hassan W, Somaily HH, Ejaz SR, Abass RR, Jasem H, Hachim SK, Adhab AH, Abood ES, Alsafari IA (2022) Synergistic effect of do** and nanotechnology to fabricate highly efficient photocatalyst for environmental remediation. J Alloys Compd 920:165876. https://doi.org/10.1016/j.jallcom.2022.165876
Wang Z, Wang P, Hu W (2022) “Clean” do** to advance 2D material phototransistors. Light Sci Appl 11:169. https://doi.org/10.1038/s41377-022-00842-4
Wang X, Sun G, Routh P, Kim D-H, Huang W, Chen P (2014) Heteroatom-doped graphene materials: syntheses, properties and applications. Chem Soc Rev 43:7067–7098. https://doi.org/10.1039/C4CS00141A
Ayala P, Arenal R, Rümmeli M, Rubio A, Pichler T (2010) The do** of carbon nanotubes with nitrogen and their potential applications. Carbon 48:575–586. https://doi.org/10.1016/j.carbon.2009.10.009
Scaccabarozzi AD, Basu A, Aniés F, Liu J, Zapata-Arteaga O, Warren R, Firdaus Y, Nugraha MI, Lin Y, Campoy-Quiles M, Koch N, Müller C, Tsetseris L, Heeney M, Anthopoulos TD (2022) Do** approaches for organic semiconductors. Chem Rev 122:4420–4492. https://doi.org/10.1021/acs.chemrev.1c00581
Zhao W, Ding J, Zou Y, Di C-a, Zhu D (2020) Chemical do** of organic semiconductors for thermoelectric applications. Chem Soc Rev 49:7210–7228. https://doi.org/10.1039/D0CS00204F
Moon SY, Gwag EH, Park JY (2018) Hydrogen generation on metal/mesoporous oxides: the effects of hierarchical structure, do**, and co-catalysts. Energ Technol 6:459–469. https://doi.org/10.1002/ente.201700409
Gwag EH, Moon SY, Mondal I, Park JY (2019) Influence of carbon do** concentration on photoelectrochemical activity of TiO 2 nanotube arrays under water oxidation. Catal Sci Technol 9:688–694. https://doi.org/10.1039/C8CY02226G
Zhao D, Dong C-L, Wang B, Chen C, Huang Y-C, Diao Z, Li S, Guo L, Shen S (2019) Synergy of dopants and defects in graphitic carbon nitride with exceptionally modulated band structures for efficient photocatalytic oxygen evolution. Adv Mater 31:1903545. https://doi.org/10.1002/adma.201903545
Cao X, Shen J, Li X-F, Luo Y (2020) Spin polarization-induced facile dioxygen activation in boron-doped graphitic carbon nitride. ACS Appl Mater Interfaces 12:52741–52748. https://doi.org/10.1021/acsami.0c16216
Agrawal S, Vasenko AS, Trivedi DJ, Prezhdo OV (2022) Charge carrier nonadiabatic dynamics in non-metal doped graphitic carbon nitride. J Chem Phys 156:094702. https://doi.org/10.1063/5.0079085
Liu Y, Zhao S, Zhang C, Fang J, ** graphitic carbon nitride with adjustable structure for highly enhanced photocatalytic hydrogen production. Carbon 182:287–296. https://doi.org/10.1016/j.carbon.2021.06.008
Zhang L, ** Z, Huang S, Huang X, Xu B, Hu L, Cui H, Ruan S, Zeng Y-J (2019) Bio-inspired carbon doped graphitic carbon nitride with booming photocatalytic hydrogen evolution. Appl Catal B 246:61–71. https://doi.org/10.1016/j.apcatb.2019.01.040
Zhao S, Liu Y, Wang Y, **e L, Fang J, Zhang Y, Zhou Y, Zhuo S (2021) C-rich graphitic carbon nitride with cross pore channels: a visible-light-driven photocatalyst for water splitting. ACS Appl Energy Mater 4:1784–1792. https://doi.org/10.1021/acsaem.0c02924
Fang J, Fan H, Li M, Long C (2015) Nitrogen self-doped graphitic carbon nitride as efficient visible light photocatalyst for hydrogen evolution. J Mater Chem A 3:13819–13826. https://doi.org/10.1039/C5TA02257F
Li J, Shen B, Hong Z, Lin B, Gao B, Chen Y (2012) A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem Commun 48:12017–12019. https://doi.org/10.1039/C2CC35862J
Shang Y, Wang Y, Lv C, **g F, Liu T, Li W, Liu S, Chen G (2022) A broom-like tube-in-tube bundle O-doped graphitic carbon nitride nanoreactor that promotes photocatalytic hydrogen evolution. Chem Eng J 431:133898. https://doi.org/10.1016/j.cej.2021.133898
Cui J, Liang S, Wang X, Zhang J (2015) First principle modeling of oxygen-doped monolayer graphitic carbon nitride. Mater Chem Phys 161:194–200. https://doi.org/10.1016/j.matchemphys.2015.05.036
Du L, Tian Q, Zheng X, Guo W, Liu W, Zhou Y, Shi F, Xu Q (2022) Supercritical CO2-tailored 2D oxygen-doped amorphous carbon nitride for enhanced photocatalytic activity. Energy Environ Sci 5:912–917. https://doi.org/10.1002/eem2.12209
Liu J, Ding G, Yu J, Liu X, Zhang X, Guo J, Ren W, Zhang J, Che R (2019) Hydrogen peroxide-assisted synthesis of oxygen-doped carbon nitride nanorods for enhanced photocatalytic hydrogen evolution. RSC Adv 9:28421–28431. https://doi.org/10.1039/C9RA04418C
Zhong K, Zhu X, Yang J, Mo Z, Qian J, He M, Song Y, Liu J, Chen H, Li H, Xu H (2022) Ultrathin structure of oxygen doped carbon nitride for efficient CO2 photocatalytic reduction. Nanotechnology 33:115404. https://doi.org/10.1088/1361-6528/ac3949
Zhang Y, Mori T, Ye J, Antonietti M (2010) Phosphorus-doped carbon nitride solid: enhanced electrical conductivity and photocurrent generation. J Am Chem Soc 132:6294–6295. https://doi.org/10.1021/ja101749y
Ran J, Ma TY, Gao G, Du X-W, Qiao SZ (2015) Porous P-doped graphitic carbon nitride nanosheets for synergistically enhanced visible-light photocatalytic H2 production. Energy Environ Sci 8:3708–3717. https://doi.org/10.1039/C5EE02650D
Guo S, Deng Z, Li M, Jiang B, Tian C, Pan Q, Fu H (2016) Phosphorus-doped carbon nitride tubes with a layered micro-nanostructure for enhanced visible-light photocatalytic hydrogen evolution. Angew Chem 128:1862–1866. https://doi.org/10.1002/ange.201508505
Guo S, Deng Z, Li M, Jiang B, Tian C, Pan Q, Fu H (2016) Phosphorus-doped carbon nitride tubes with a layered micro-nanostructure for enhanced visible-light photocatalytic hydrogen evolution. Angew Chem Int Ed 55:1830–1834. https://doi.org/10.1002/anie.201508505
Zhu Y-P, Ren T-Z, Yuan Z-Y (2015) Mesoporous phosphorus-doped g-C3N4 nanostructured flowers with superior photocatalytic hydrogen evolution performance. ACS Appl Mater Interfaces 7:16850–16856. https://doi.org/10.1021/acsami.5b04947
Liu B, Ye L, Wang R, Yang J, Zhang Y, Guan R, Tian L, Chen X (2018) Phosphorus-doped graphitic carbon nitride nanotubes with amino-rich surface for efficient CO2 capture, enhanced photocatalytic activity, and product selectivity. ACS Appl Mater Interfaces 10:4001–4009. https://doi.org/10.1021/acsami.7b17503
Fiss BG, Douglas G, Ferguson M, Becerra J, Valdez J, Do T-O, Friščić T, Moores A (2022) Mechanosynthesis of a structurally characterized, well-defined graphitic phosphorus-linked carbon nitride (g-PCN) with water splitting activity. Adv Mater Interfaces 9:2201555. https://doi.org/10.1002/admi.202201555
Babu P, Kim H, Park JY, Naik B (2022) Trioctylphosphine oxide (TOPO)-assisted facile fabrication of phosphorus-incorporated nanostructured carbon nitride toward photoelectrochemical water splitting with enhanced activity. Inorg Chem 61:1368–1376. https://doi.org/10.1021/acs.inorgchem.1c02863
He F, Chen G, Yu Y, Zhou Y, Zheng Y, Hao S (2015) The sulfur-bubble template-mediated synthesis of uniform porous g-C3N4 with superior photocatalytic performance. Chem Commun 51:425–427. https://doi.org/10.1039/C4CC07106A
Lv H, Huang Y, Koodali RT, Liu G, Zeng Y, Meng Q, Yuan M (2020) Synthesis of sulfur-doped 2D graphitic carbon nitride nanosheets for efficient photocatalytic degradation of phenol and hydrogen evolution. ACS Appl Mater Interfaces 12:12656–12667. https://doi.org/10.1021/acsami.9b19057
Wang Y, Di Y, Antonietti M, Li H, Chen X, Wang X (2010) Excellent visible-light photocatalysis of fluorinated polymeric carbon nitride solids. Chem Mater 22:5119–5121. https://doi.org/10.1021/cm1019102
Liu C, Zhang Y, Dong F, Reshak AH, Ye L, Pinna N, Zeng C, Zhang T, Huang H (2017) Chlorine intercalation in graphitic carbon nitride for efficient photocatalysis. Appl Catal B 203:465–474. https://doi.org/10.1016/j.apcatb.2016.10.002
Lan Z-A, Zhang G, Wang X (2016) A facile synthesis of Br-modified g-C3N4 semiconductors for photoredox water splitting. Appl Catal B 192:116–125. https://doi.org/10.1016/j.apcatb.2016.03.062
Han Q, Hu C, Zhao F, Zhang Z, Chen N, Qu L (2015) One-step preparation of iodine-doped graphitic carbon nitride nanosheets as efficient photocatalysts for visible light water splitting. J Mater Chem A 3:4612–4619. https://doi.org/10.1039/C4TA06093H
Zhu B, Zhang J, Jiang C, Cheng B, Yu J (2017) First principle investigation of halogen-doped monolayer g-C3N4 photocatalyst. Appl Catal B 207:27–34. https://doi.org/10.1016/j.apcatb.2017.02.020
Cheon JY, Kim JH, Kim JH, Goddeti KC, Park JY, Joo SH (2014) Intrinsic relationship between enhanced oxygen reduction reaction activity and nanoscale work function of doped carbons. J Am Chem Soc 136:8875–8878. https://doi.org/10.1021/ja503557x
Mondal I, Mahata A, Kim H, Pal U, De Angelis F, Park JY (2020) A combined experimental and theoretical approach revealing a direct mechanism for bifunctional water splitting on doped copper phosphide. Nanoscale 12:17769–17779. https://doi.org/10.1039/D0NR03414B
Samokhvalov A (2017) Hydrogen by photocatalysis with nitrogen codoped titanium dioxide. Renew Sust Energ Rev 72:981–1000. https://doi.org/10.1016/j.rser.2017.01.024
Ye Z, He H, Jiang L (2018) Co-do**: an effective strategy for achieving stable p-type ZnO thin films. Nano Energy 52:527–540. https://doi.org/10.1016/j.nanoen.2018.08.001
Gao K, Wang B, Tao L, Cunning BV, Zhang Z, Wang S, Ruoff RS, Qu L (2019) Efficient metal-free electrocatalysts from N-doped carbon nanomaterials: mono-do** and co-do**. Adv Mater 31:1805121. https://doi.org/10.1002/adma.201805121
Poornaprakash B, Chalapathi U, Kumar M, Vattikuti SVP, Rajitha B, Poojitha PT, Park S-H (2021) Tailoring the optical and magnetic properties of ZnS nanoparticles via 3d and 4f elements co-do**. Mater Sci Semicond Process 121:105395. https://doi.org/10.1016/j.mssp.2020.105395
Obeid MM, Stampfl C, Bafekry A, Guan Z, Jappor HR, Nguyen CV, Naseri M, Hoat DM, Hieu NN, Krauklis AE, Vu TV, Gogova D (2020) First-principles investigation of nonmetal doped single-layer BiOBr as a potential photocatalyst with a low recombination rate. Phys Chem Chem Phys 22:15354–15364. https://doi.org/10.1039/D0CP02007A
Wu H-C, Lin S-W, Wu J-S (2012) Effects of nitrogen concentration on N-doped anatase TiO2: density functional theory and Hubbard U analysis. J Alloys Compd 522:46–50. https://doi.org/10.1016/j.jallcom.2012.01.071
Wang K, Fu J, Zheng Y (2019) Insights into photocatalytic CO2 reduction on C3N4: strategy of simultaneous B, K co-do** and enhancement by N vacancies. Appl Catal B 254:270–282. https://doi.org/10.1016/j.apcatb.2019.05.002
Sun L, Zhao X, Cheng X, Sun H, Li Y, Li P, Fan W (2011) Evaluating the C, N, and F pairwise codo** effect on the enhanced photoactivity of ZnWO4: the charge compensation mechanism in donor–acceptor pairs. J Phys Chem C 115:15516–15524. https://doi.org/10.1021/jp204324n
Pu K, Dai X, Jia D, Tao W, Liu F, Zhang X, Song J, Zhao T, Hao Y (2020) Structural, electrical and optical properties of halogen doped phosphorene based on density functional theory. J Alloys Compd 812:152125. https://doi.org/10.1016/j.jallcom.2019.152125
Boshir Ahmed M, Alom J, Hasan MS, Asaduzzaman M, Rahman MS, Hossen R, Abu Hasan Johir M, Taufiq Alam M, Zhou JL, Zhu Y, Zargar M (2023) General do** chemistry of carbon materials. ChemNanoMat 9:e202200482. https://doi.org/10.1002/cnma.202200482
Sun H, Zhao X, Zhang L, Fan W (2011) Origin of the enhanced visible photocatalytic activity in (N, C)-codoped ZnS studied from density functional theory. J Phys Chem C 115:2218–2227. https://doi.org/10.1021/jp110263e
Yu S-S, Zheng W-T (2010) Effect of N/B do** on the electronic and field emission properties for carbon nanotubes, carbon nanocones, and graphene nanoribbons. Nanoscale 2:1069–1082. https://doi.org/10.1039/C0NR00002G
Zunger A, Malyi OI (2021) Understanding do** of quantum materials. Chem Rev 121:3031–3060. https://doi.org/10.1021/acs.chemrev.0c00608
Babu P, Mohanty S, Naik B, Parida K (2018) Synergistic dffects of boron and sulfur co-do** into graphitic carbon nitride framework for enhanced photocatalytic activity in visible light driven hydrogen generation. ACS Appl Energy Mater 1:5936–5947. https://doi.org/10.1021/acsaem.8b00956
Wu J, Li N, Zhang X-H, Fang H-B, Zheng Y-Z, Tao X (2018) Heteroatoms binary-doped hierarchical porous g-C3N4 nanobelts for remarkably enhanced visible-light-driven hydrogen evolution. Appl Catal B 226:61–70. https://doi.org/10.1016/j.apcatb.2017.12.045
Song X, Li X, Zhang X, Wu Y, Ma C, Huo P, Yan Y (2020) Fabricating C and O co-doped carbon nitride with intramolecular donor-acceptor systems for efficient photoreduction of CO2 to CO. Appl Catal B 268:118736. https://doi.org/10.1016/j.apcatb.2020.118736
Kumar S, Gawande MB, Kopp J, Kment S, Varma RS, Zbořil R (2020) P- and F-co-doped carbon nitride nanocatalysts for photocatalytic CO2 reduction and thermocatalytic furanics synthesis from sugars. Chemsuschem 13:5231–5238. https://doi.org/10.1002/cssc.202001172
Liang Q, Li Z, Huang Z-H, Kang F, Yang Q-H (2015) Holey graphitic carbon nitride nanosheets with carbon vacancies for highly improved photocatalytic hydrogen production. Adv Funct Mater 25:6885–6892. https://doi.org/10.1002/adfm.201503221
Shen M, Zhang L, Wang M, Tian J, ** X, Guo L, Wang L, Shi J (2019) Carbon-vacancy modified graphitic carbon nitride: enhanced CO2 photocatalytic reduction performance and mechanism probing. J Mater Chem A 7:1556–1563. https://doi.org/10.1039/C8TA09302D
Li Y, Zhong J, Li J (2022) Rich carbon vacancies facilitated solar light-driven photocatalytic hydrogen generation over g-C3N4 treated in H2 atmosphere. Int J Hydrogen Energy 47:39886–39897. https://doi.org/10.1016/j.ijhydene.2022.09.147
Tu W, Xu Y, Wang J, Zhang B, Zhou T, Yin S, Wu S, Li C, Huang Y, Zhou Y, Zou Z, Robertson J, Kraft M, Xu R (2017) Investigating the role of tunable nitrogen vacancies in graphitic carbon nitride nanosheets for efficient visible-light-driven H2 evolution and CO2 reduction. ACS Sustain Chem Eng 5:7260–7268. https://doi.org/10.1021/acssuschemeng.7b01477
Yu H, Shi R, Zhao Y, Bian T, Zhao Y, Zhou C, Waterhouse GIN, Wu L-Z, Tung C-H, Zhang T (2017) Alkali-assisted synthesis of nitrogen deficient graphitic carbon nitride with tunable band structures for efficient visible-light-driven hydrogen evolution. Adv Mater 29:1605148. https://doi.org/10.1002/adma.201605148
Niu P, Qiao M, Li Y, Huang L, Zhai T (2018) Distinctive defects engineering in graphitic carbon nitride for greatly extended visible light photocatalytic hydrogen evolution. Nano Energy 44:73–81. https://doi.org/10.1016/j.nanoen.2017.11.059
Huang Y, Liu J, Zhao C, Jia X, Ma M, Qian Y, Yang C, Liu K, Tan F, Wang Z, Li X, Qu S, Wang Z (2020) Facile synthesis of defect-modified thin-layered and porous g-C3N4 with synergetic improvement for photocatalytic H2 production. ACS Appl Mater Interfaces 12:52603–52614. https://doi.org/10.1021/acsami.0c14262
Dias EM, Christoforidis KC, Francàs L, Petit C (2018) Tuning thermally treated graphitic carbon nitride for H2 evolution and CO2 photoreduction: the effects of material properties and mid-gap States. ACS Appl Energy Mater 1:6524–6534. https://doi.org/10.1021/acsaem.8b01441
Tang J-Y, Kong XY, Ng B-J, Chew Y-H, Mohamed AR, Chai S-P (2019) Midgap-state-mediated two-step photoexcitation in nitrogen defect-modified g-C3N4 atomic layers for superior photocatalytic CO2 reduction. Catal Sci Technol 9:2335–2343. https://doi.org/10.1039/C9CY00449A
Park JY, Baker LR, Somorjai GA (2015) Role of hot electrons and metal–oxide interfaces in surface chemistry and catalytic reactions. Chem Rev 115:2781–2817. https://doi.org/10.1021/cr400311p
Park JY, Kim SM, Lee H, Naik B (2014) Hot electron and surface plasmon-driven catalytic reaction in metal–semiconductor nanostructures. Catal Lett 144:1996–2004. https://doi.org/10.1007/s10562-014-1333-2
Zhang Z, Yates JT Jr (2012) Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces. Chem Rev 112:5520–5551. https://doi.org/10.1021/cr3000626
Jiao Y, Hellman A, Fang Y, Gao S, Käll M (2015) Schottky barrier formation and band bending revealed by first- principles calculations. Sci Rep 5:11374. https://doi.org/10.1038/srep11374
Ozawa K, Emori M, Yamamoto S, Yukawa R, Yamamoto S, Hobara R, Fujikawa K, Sakama H, Matsuda I (2014) Electron–hole recombination time at TiO2 single-crystal surfaces: Influence of surface band bending. J Phys Chem Lett 5:1953–1957. https://doi.org/10.1021/jz500770c
Wang C, Astruc D (2014) Nanogold plasmonic photocatalysis for organic synthesis and clean energy conversion. Chem Soc Rev 43:7188–7216. https://doi.org/10.1039/C4CS00145A
Christopher P, **n H, Marimuthu A, Linic S (2012) Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat Mater 11:1044–1050. https://doi.org/10.1038/nmat3454
Linic S, Christopher P, Ingram DB (2011) Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat Mater 10:911–921. https://doi.org/10.1038/nmat3151
Clavero C (2014) Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat Photonics 8:95–103. https://doi.org/10.1038/nphoton.2013.238
Li K, Hogan NJ, Kale MJ, Halas NJ, Nordlander P, Christopher P (2017) Balancing near-field enhancement, absorption, and scattering for effective antenna–reactor plasmonic photocatalysis. Nano Lett 17:3710–3717. https://doi.org/10.1021/acs.nanolett.7b00992
Marchuk K, Willets KA (2014) Localized surface plasmons and hot electrons. Chem Phys 445:95–104. https://doi.org/10.1016/j.chemphys.2014.10.016
Zhao J, Xue S, Ji R, Li B, Li J (2021) Localized surface plasmon resonance for enhanced electrocatalysis. Chem Soc Rev 50:12070–12097. https://doi.org/10.1039/D1CS00237F
Hou W, Cronin SB (2013) A review of surface plasmon resonance-enhanced photocatalysis. Adv Funct Mater 23:1612–1619. https://doi.org/10.1002/adfm.201202148
Park JY, Kim SM, Lee H, Nedrygailov II (2015) Hot-electron-mediated surface chemistry: toward electronic control of catalytic activity. Acc Chem Res 48:2475–2483. https://doi.org/10.1021/acs.accounts.5b00170
Lee SW, Jeon B, Lee H, Park JY (2022) Hot electron phenomena at solid–liquid interfaces. J Phys Chem Lett 13:9435–9448. https://doi.org/10.1021/acs.jpclett.2c02319
Kim SM, Lee SW, Moon SY, Park JY (2016) The effect of hot electrons and surface plasmons on heterogeneous catalysis. J Phys: Condens Matter 28:254002. https://doi.org/10.1088/0953-8984/28/25/254002
Lee H, Lee H, Park JY (2019) Direct imaging of surface plasmon-driven hot electron flux on the Au nanoprism/TiO2. Nano Lett 19:891–896. https://doi.org/10.1021/acs.nanolett.8b04119
Mondal I, Lee H, Kim H, Park JY (2020) Plasmon-induced hot carrier separation across dual interface in gold–nickel phosphide heterojunction for photocatalytic water splitting. Adv Funct Mater 30:1908239. https://doi.org/10.1002/adfm.201908239
Ueno K, Misawa H (2013) Surface plasmon-enhanced photochemical reactions. J Photochem Photobiol C 15:31–52. https://doi.org/10.1016/j.jphotochemrev.2013.04.001
Sayed M, Yu J, Liu G, Jaroniec M (2022) Non-noble plasmonic metal-based photocatalysts. Chem Rev 122:10484–10537. https://doi.org/10.1021/acs.chemrev.1c00473
Zhou M, Li C, Fang J (2021) Noble-metal based random alloy and intermetallic nanocrystals: syntheses and applications. Chem Rev 121:736–795. https://doi.org/10.1021/acs.chemrev.0c00436
Hartland GV (2011) Optical studies of dynamics in noble metal nanostructures. Chem Rev 111:3858–3887. https://doi.org/10.1021/cr1002547
Wang M, Ye M, Iocozzia J, Lin C, Lin Z (2016) Plasmon-mediated solar energy conversion via photocatalysis in noble metal/semiconductor composites. Adv Sci 3:1600024. https://doi.org/10.1002/advs.201600024
Jain PK, El-Sayed MA (2010) Plasmonic coupling in noble metal nanostructures. Chem Phys Lett 487:153–164. https://doi.org/10.1016/j.cplett.2010.01.062
Fan Z, Zhang H (2016) Crystal phase-controlled synthesis, properties and applications of noble metal nanomaterials. Chem Soc Rev 45:63–82. https://doi.org/10.1039/C5CS00467E
Song K, Lee H, Lee M, Park JY (2021) Plasmonic hot hole-driven water splitting on Au nanoprisms/p-type GaN. ACS Energy Lett 6:1333–1339. https://doi.org/10.1021/acsenergylett.1c00366
DuChene JS, Tagliabue G, Welch AJ, Cheng W-H, Atwater HA (2018) Hot hole collection and photoelectrochemical CO2 reduction with plasmonic Au/p-GaN photocathodes. Nano Lett 18:2545–2550. https://doi.org/10.1021/acs.nanolett.8b00241
Lee H, Park Y, Song K, Park JY (2022) Surface plasmon-induced hot carriers: generation, detection, and applications. Acc Chem Res 55:3727–3737. https://doi.org/10.1021/acs.accounts.2c00623
Yuan L, Lou M, Clark BD, Lou M, Zhou L, Tian S, Jacobson CR, Nordlander P, Halas NJ (2020) Morphology-dependent reactivity of a plasmonic photocatalyst. ACS Nano 14:12054–12063. https://doi.org/10.1021/acsnano.0c05383
Lee SW, Lee H, Park Y, Kim H, Somorjai GA, Park JY (2021) Surface chemistry of hot electron and metal-oxide interfaces. Surf Sci Rep 76:100532. https://doi.org/10.1016/j.surfrep.2021.100532
Bai J, Lu B, Han Q, Li Q, Qu L (2018) (111) Facets-oriented Au-decorated carbon nitride nanoplatelets for visible-light-driven overall water splitting. ACS Appl Mater Interfaces 10:38066–38072. https://doi.org/10.1021/acsami.8b13371
Li F, Zhou H, Fan J, **ang Q (2020) Amine-functionalized graphitic carbon nitride decorated with small-sized Au nanoparticles for photocatalytic CO2 reduction. J Colloid Interface Sci 570:11–19. https://doi.org/10.1016/j.jcis.2020.02.108
Chen L, Li H, Li H, Li H, Qi W, Zhang Q, Zhu J, Zhao P, Yang S (2022) Accelerating photogenerated charge kinetics via the g-C3N4 Schottky junction for enhanced visible-light-driven CO2 reduction. Appl Catal B 318:121863. https://doi.org/10.1016/j.apcatb.2022.121863
Zhao D, Wang Y, Dong C-L, Huang Y-C, Chen J, Xue F, Shen S, Guo L (2021) Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting. Nat Energy 6:388–397. https://doi.org/10.1038/s41560-021-00795-9
Thaweesak S, Wang S, Lyu M, **ao M, Peerakiatkhajohn P, Wang L (2017) Boron-doped graphitic carbon nitride nanosheets for enhanced visible light photocatalytic water splitting. Dalton Trans 46:10714–10720. https://doi.org/10.1039/C7DT00933J
Qi K, Cui N, Zhang M, Ma Y, Wang G, Zhao Z, Khataee A (2021) Ionic liquid-assisted synthesis of porous boron-doped graphitic carbon nitride for photocatalytic hydrogen production. Chemosphere 272:129953. https://doi.org/10.1016/j.chemosphere.2021.129953
Zhao S, Zhang C, Wang Y, Zhao K, Zhou Y, Li L, Zhuo S (2023) Facile construction of carbon doped carbon nitride tube with increased π-electron density for highly efficient hydrogen production. Colloids Surf A Physicochem Eng Asp 660:130872. https://doi.org/10.1016/j.colsurfa.2022.130872
Huang Y, Li D, Fang Z, Chen R, Luo B, Shi W (2019) Controlling carbon self-do** site of g-C3N4 for highly enhanced visible-light-driven hydrogen evolution. Appl Catal B 254:128–134. https://doi.org/10.1016/j.apcatb.2019.04.082
Jiang W, Ruan Q, **e J, Chen X, Zhu Y, Tang J (2018) Oxygen-doped carbon nitride aerogel: a self-supported photocatalyst for solar-to-chemical energy conversion. Appl Catal B 236:428–435. https://doi.org/10.1016/j.apcatb.2018.05.050
Zhang Y, Chen Z, Li J, Lu Z, Wang X (2021) Self-assembled synthesis of oxygen-doped g-C3N4 nanotubes in enhancement of visible-light photocatalytic hydrogen. J Energy Chem 54:36–44. https://doi.org/10.1016/j.jechem.2020.05.043
Chen Y, Liu X, Hou L, Guo X, Fu R, Sun J (2020) Construction of covalent bonding oxygen-doped carbon nitride/graphitic carbon nitride Z-scheme heterojunction for enhanced visible-light-driven H2 evolution. Chem Eng J 383:123132. https://doi.org/10.1016/j.cej.2019.123132
Pandi K, Rajan A, Dhileepan MD, Sekar K, Neppolian B (2022) Enhanced solar hydrogen production by template-free oxygen doped porous graphitic carbon nitride photocatalysts. Mater Today Chem 26:101173. https://doi.org/10.1016/j.mtchem.2022.101173
Wen J, Zhang S, Liu Y, Zhai Y (2022) Formic acid assisted fabrication of Oxygen-doped Rod-like carbon nitride with improved photocatalytic hydrogen evolution. J Colloid Interface Sci 624:338–347. https://doi.org/10.1016/j.jcis.2022.05.130
Zhang G, Xu Y, He C, Zhang P, Mi H (2021) Oxygen-doped crystalline carbon nitride with greatly extended visible-light-responsive range for photocatalytic H2 generation. Appl Catal B 283:119636. https://doi.org/10.1016/j.apcatb.2020.119636
Fang X-X, Ma L-B, Liang K, Zhao S-J, Jiang Y-F, Ling C, Zhao T, Cheang T-Y, Xu A-W (2019) The do** of phosphorus atoms into graphitic carbon nitride for highly enhanced photocatalytic hydrogen evolution. J Mater Chem A 7:11506–11512. https://doi.org/10.1039/C9TA01646E
Wang M, Qin Z, Diao Z, Li R, Zhong J, Ma D, Chen Y (2020) Red phosphorus/carbon nitride van der Waals heterostructure for photocatalytic pure water splitting under wide-spectrum light irradiation. ACS Sustain Chem Eng 8:13459–13466. https://doi.org/10.1021/acssuschemeng.0c04372
Zhu Y, Li J, Cao J, Lv C, Huang G, Zhang G, Xu Y, Zhang S, Meng P, Zhan T, Yang D (2020) Phosphorus-doped polymeric carbon nitride nanosheets for enhanced photocatalytic hydrogen production. APL Mater 8:041108. https://doi.org/10.1063/1.5143711
Li B, Si Y, Fang Q, Shi Y, Huang W-Q, Hu W, Pan A, Fan X, Huang G-F (2020) Hierarchical self-assembly of well-defined Louver-like P-doped carbon nitride nanowire arrays with highly efficient hydrogen evolution. Nanomicro Lett 12:52. https://doi.org/10.1007/s40820-020-0399-1
Wang C, Yang C, Qin J, Rajendran S, Zhang X (2022) A facile template synthesis of phosphorus-doped graphitic carbon nitride hollow structures with high photocatalytic hydrogen production activity. Mater Chem Phys 275:125299. https://doi.org/10.1016/j.matchemphys.2021.125299
Wang H, Bian Y, Hu J, Dai L (2018) Highly crystalline sulfur-doped carbon nitride as photocatalyst for efficient visible-light hydrogen generation. Appl Catal B 238:592–598. https://doi.org/10.1016/j.apcatb.2018.07.023
Hong J, ** for enhanced photocatalytic hydrogen evolution from water under visible light. J Mater Chem 22:15006–15012. https://doi.org/10.1039/C2JM32053C
Fei T, Qin C, Zhang Y, Dong G, Wang Y, Zhou Y, Cui M (2021) A 3D peony-like sulfur-doped carbon nitride synthesized by self-assembly for efficient photocatalytic hydrogen production. Int J Hydrogen Energy 46:20481–20491. https://doi.org/10.1016/j.ijhydene.2021.03.148
Du J, Li S, Du Z, Meng S, Li B (2021) Boron/oxygen-codoped graphitic carbon nitride nanomesh for efficient photocatalytic hydrogen evolution. Chem Eng J 407:127114. https://doi.org/10.1016/j.cej.2020.127114
Jiang Y, Fang S, Cao C, Hong E, Zeng L, Yang W, Huang L, Yang C (2022) Enhanced light harvesting and charge separation of carbon and oxygen co-doped carbon nitride as excellent photocatalyst for hydrogen evolution reaction. J Colloid Interface Sci 612:367–376. https://doi.org/10.1016/j.jcis.2021.12.077
Wang H, Wang B, Bian Y, Dai L (2017) Enhancing photocatalytic activity of graphitic carbon nitride by codo** with P and C for efficient hydrogen generation. ACS Appl Mater Interfaces 9:21730–21737. https://doi.org/10.1021/acsami.7b02445
Yan B, Du C, Yang G (2020) Constructing built-in electric field in ultrathin graphitic carbon nitride nanosheets by N and O codo** for enhanced photocatalytic hydrogen evolution activity. Small 16:1905700. https://doi.org/10.1002/smll.201905700
Shao Y, Hao X, Lu S, ** Z (2023) Molten salt-assisted synthesis of nitrogen-vacancy crystalline graphitic carbon nitride with tunable band structures for efficient photocatalytic overall water splitting. Chem Eng J 454:140123. https://doi.org/10.1016/j.cej.2022.140123
Yang F, Li H, Pan K, Wang S, Sun H, **e Y, Xu Y, Wu J, Zhou W (2021) Engineering surface N-vacancy defects of ultrathin mesoporous carbon nitride nanosheets as efficient visible-light-driven photocatalysts. Solar RRL 5:2000610. https://doi.org/10.1002/solr.202000610
Zeng Y, Li H, Luo J, Yuan J, Wang L, Liu C, **a Y, Liu M, Luo S, Cai T, Liu S, Crittenden JC (2019) Sea-urchin-structure g-C3N4 with narrow bandgap (˜2.0 eV) for efficient overall water splitting under visible light irradiation. Appl Catal B 249:275–281. https://doi.org/10.1016/j.apcatb.2019.03.010
Nguyen C-C, Do T-O (2018) Engineering the high concentration of N3C nitrogen vacancies toward strong solar light-driven photocatalyst-based g-C3N4. ACS Appl Energy Mater 1:4716–4723. https://doi.org/10.1021/acsaem.8b00839
Zhang Y, Huang Z, Dong C-L, Shi J, Cheng C, Guan X, Zong S, Luo B, Cheng Z, Wei D, Huang Y-c, Shen S, Guo L (2022) Synergistic effect of nitrogen vacancy on ultrathin graphitic carbon nitride porous nanosheets for highly efficient photocatalytic H2 evolution. Chem Eng J 431:134101. https://doi.org/10.1016/j.cej.2021.134101
Li H, ** C, Wang Z, Liu Y, Wang P, Zheng Z, Whangbo M-H, Kou L, Li Y, Dai Y, Huang B (2019) Effect of the intra- and inter-triazine N-vacancies on the photocatalytic hydrogen evolution of graphitic carbon nitride. Chem Eng J 369:263–271. https://doi.org/10.1016/j.cej.2019.03.095
Li Q, Tong Y, Zeng Y, Gu X-K, Ding M (2022) Carbohydrate-regulated synthesis of ultrathin porous nitrogen-vacancy polymeric carbon nitride for highly efficient visible-light hydrogen evolution. Chem Eng J 450:138010. https://doi.org/10.1016/j.cej.2022.138010
Jiang Y, Sun Z, Tang C, Zhou Y, Zeng L, Huang L (2019) Enhancement of photocatalytic hydrogen evolution activity of porous oxygen doped g-C3N4 with nitrogen defects induced by changing electron transition. Appl Catal B 240:30–38. https://doi.org/10.1016/j.apcatb.2018.08.059
Wu J, Li N, Fang H-B, Li X, Zheng Y-Z, Tao X (2019) Nitrogen vacancies modified graphitic carbon nitride: Scalable and one-step fabrication with efficient visible-light-driven hydrogen evolution. Chem Eng J 358:20–29. https://doi.org/10.1016/j.cej.2018.09.208
Liang L, Shi L, Wang F (2019) Fabrication of large surface area nitrogen vacancy modified graphitic carbon nitride with improved visible-light photocatalytic performance. Diamond Relat Mater 91:230–236. https://doi.org/10.1016/j.diamond.2018.11.025
Zhang D, Guo Y, Zhao Z (2018) Porous defect-modified graphitic carbon nitride via a facile one-step approach with significantly enhanced photocatalytic hydrogen evolution under visible light irradiation. Appl Catal B 226:1–9. https://doi.org/10.1016/j.apcatb.2017.12.044
Li H, Ning F, Chen X, Shi A (2021) Effect of carbon and nitrogen double vacancies on the improved photocatalytic hydrogen evolution over porous carbon nitride nanosheets. Catal Sci Technol 11:3270–3278. https://doi.org/10.1039/D0CY02453H
Wang X, Wu L, Wang Z, Wu H, Zhou X, Ma H, Zhong H, **ng Z, Cai G, Jiang C, Ren F (2019) C/N vacancy co-enhanced visible-light-driven hydrogen evolution of g-C3N4 nanosheets through controlled He+ ion irradiation. Solar RRL 3:1800298. https://doi.org/10.1002/solr.201800298
Lin Y, Yang Y, Guo W, Wang L, Zhang R, Liu Y, Zhai Y (2021) Preparation of double-vacancy modified carbon nitride to greatly improve the activity of photocatalytic hydrogen generation. Appl Surf Sci 560:150029. https://doi.org/10.1016/j.apsusc.2021.150029
Yu W, Shan X, Zhao Z (2020) Unique nitrogen-deficient carbon nitride homojunction prepared by a facile inserting-removing strategy as an efficient photocatalyst for visible light-driven hydrogen evolution. Appl Catal B 269:118778. https://doi.org/10.1016/j.apcatb.2020.118778
Zhang Q, Chen X, Yang Z, Yu T, Liu L, Ye J (2022) Precisely tailoring nitrogen defects in carbon nitride for efficient photocatalytic overall water splitting. ACS Appl Mater Interfaces 14:3970–3979. https://doi.org/10.1021/acsami.1c19638
Shan X, Ge G, Zhao Z (2019) Fabrication of tubular g-C3N4 with N-defects and extended π-conjugated system for promoted photocatalytic hydrogen production. ChemCatChem 11:1534–1544. https://doi.org/10.1002/cctc.201801803
Wu M, Gong Y, Nie T, Zhang J, Wang R, Wang H, He B (2019) Template-free synthesis of nanocage-like g-C3N4 with high surface area and nitrogen defects for enhanced photocatalytic H2 activity. J Mater Chem A 7:5324–5332. https://doi.org/10.1039/C8TA12076E
Fang H-X, Guo H, Niu C-G, Liang C, Huang D-W, Tang N, Liu H-Y, Yang Y-Y, Li L (2020) Hollow tubular graphitic carbon nitride catalyst with adjustable nitrogen vacancy: enhanced optical absorption and carrier separation for improving photocatalytic activity. Chem Eng J 402:126185. https://doi.org/10.1016/j.cej.2020.126185
Ruan D, Kim S, Fujitsuka M, Majima T (2018) Defects rich g-C3N4 with mesoporous structure for efficient photocatalytic H2 production under visible light irradiation. Appl Catal B 238:638–646. https://doi.org/10.1016/j.apcatb.2018.07.028
Liu J, Fang W, Wei Z, Qin Z, Jiang Z, Shangguan W (2018) Efficient photocatalytic hydrogen evolution on N-deficient g-C3N4 achieved by a molten salt post-treatment approach. Appl Catal B 238:465–470. https://doi.org/10.1016/j.apcatb.2018.07.021
Ge G, Guo X, Song C, Zhao Z (2018) Reconstructing supramolecular aggregates to nitrogen-deficient g-C3N4 bunchy tubes with enhanced photocatalysis for H2 production. ACS Appl Mater Interfaces 10:18746–18753. https://doi.org/10.1021/acsami.8b04227
**g L, Wang D, Xu Y, **e M, Yan J, He M, Song Z, Xu H, Li H (2020) Porous defective carbon nitride obtained by a universal method for photocatalytic hydrogen production from water splitting. J Colloid Interface Sci 566:171–182. https://doi.org/10.1016/j.jcis.2020.01.044
Wang B, Cai H, Zhao D, Song M, Guo P, Shen S, Li D, Yang S (2019) Enhanced photocatalytic hydrogen evolution by partially replaced corner-site C atom with P in g-C3N4. Appl Catal B 244:486–493. https://doi.org/10.1016/j.apcatb.2018.10.044
Wang B, Bai C, Wang Z, She P, Sun H, Lu G, Liang S, Liu Z (2022) Phosphorus–oxygen-codoped graphitic carbon nitride for enhanced hydrogen evolution and photocatalytic degradation under visible light irradiation. ACS Appl Energy Mater 5:5774–5784. https://doi.org/10.1021/acsaem.2c00099
Mo R, Li J, Tang Y, Li H, Zhong J (2019) Introduction of nitrogen defects into a graphitic carbon nitride framework by selenium vapor treatment for enhanced photocatalytic hydrogen production. Appl Surf Sci 476:552–559. https://doi.org/10.1016/j.apsusc.2019.01.085
Liu J, Wei Z, Fang W, Jiang Z, Shangguan W (2019) Enhanced photocatalytic hydrogen evolution of the hydrogenated deficient g-C3N4 via surface hydrotreating. ChemCatChem 11:6275–6281. https://doi.org/10.1002/cctc.201900284
Yang P, Ou H, Fang Y, Wang X (2017) A facile steam reforming strategy to delaminate layered carbon nitride semiconductors for photoredox catalysis. Angew Chem Int Ed 56:3992–3996. https://doi.org/10.1002/anie.201700286
Zhang L, Wang A, Zhang M, ** Z, Yi H, Wang M (2021) Effective photocatalytic hydrogen evolution using covalent triazine framework-derived carbon nitride nanofiber containing carbon vacancies for visible-light-driven. Appl Sci 11:7222. https://doi.org/10.3390/app11167222
Zhao X, Yi X, Pan W, Wang Y, Luo S, Zhang Y, ** in graphitic carbon nitride with suppressed deep trap** for enhancement in CO2 photoreduction activity. J Mater Sci Technol 133:135–144. https://doi.org/10.1016/j.jmst.2022.06.022
Wang H-z, Zhao Y-z, Yang Z-x, Bi X-z, Wang Z-l, Wu M-b (2022) Oxygen-incorporated carbon nitride porous nanosheets for highly efficient photoelectrocatalytic CO2 reduction to formate. New Carbon Mater 37:1135–1142. https://doi.org/10.1016/S1872-5805(22)60619-X
Guo Y, Wang M, Tian J, Shen M, Zhang L, Shi J (2020) Probing the effect of P-do** in polymeric carbon nitride on CO2 photocatalytic reduction. Dalton Trans 49:15750–15757. https://doi.org/10.1039/D0DT03229H
Zeng D, Wang X, Liu Y, Liu D, Zhang Z, Fei L, Robertson J, Kuai C, Guo Y (2022) Self-poisoning by C2 products in CO2 photoreduction using a phosphorus-doped carbon nitride with nitrogen vacancies. ACS Sustain Chem Eng 10:5758–5769. https://doi.org/10.1021/acssuschemeng.1c07992
Chen M, Guo M, Zhai M, Xu J, Wang L (2023) Manipulating electronic structure and light absorption of carbon nitride via P-do** and local crystallization for efficient photocatalytic reduction of CO2. J CO2 Util 68:102392. https://doi.org/10.1016/j.jcou.2023.102392
Huang X, Gu W, Hu S, Hu Y, Zhou L, Lei J, Wang L, Liu Y, Zhang J (2020) Phosphorus-doped inverse opal g-C3N4 for efficient and selective CO generation from photocatalytic reduction of CO2. Catal Sci Technol 10:3694–3700. https://doi.org/10.1039/D0CY00457J
Wang W-F, Qiu L-Q, Chen K-H, Li H-R, Feng L-F, He L-N (2022) Morphology and element do** effects: phosphorus-doped hollow polygonal g-C3N4 rods for visible light-driven CO2 reduction. New J Chem 46:3017–3025. https://doi.org/10.1039/D1NJ05736G
Shcherban ND, Filonenko SM, Ovcharov ML, Mishura AM, Skoryk MA, Aho A, Murzin DY (2016) Simple method for preparing of sulfur–doped graphitic carbon nitride with superior activity in CO2 photoreduction. ChemistrySelect 1:4987–4993. https://doi.org/10.1002/slct.201601283
Wang K, Li Q, Liu B, Cheng B, Ho W, Yu J (2015) Sulfur-doped g-C3N4 with enhanced photocatalytic CO2-reduction performance. Appl Catal B 176–177:44–52. https://doi.org/10.1016/j.apcatb.2015.03.045
Arumugam M, Tahir M, Praserthdam P (2022) Effect of nonmetals (B, O, P, and S) doped with porous g-C3N4 for improved electron transfer towards photocatalytic CO2 reduction with water into CH4. Chemosphere 286:131765. https://doi.org/10.1016/j.chemosphere.2021.131765
Zhu Z, Liu Z, Tang X, Reeti K, Huo P, Wong JW-C, Zhao J (2021) Sulfur-doped g-C3N4 for efficient photocatalytic CO2 reduction: insights by experiment and first-principles calculations. Catal Sci Technol 11:1725–1736. https://doi.org/10.1039/D0CY02382E
Yang P, Zhuzhang H, Wang R, Lin W, Wang X (2019) Carbon vacancies in a melon polymeric matrix promote photocatalytic carbon dioxide conversion. Angew Chem Int Ed 58:1134–1137. https://doi.org/10.1002/anie.201810648
Shi H, Long S, Hou J, Ye L, Sun Y, Ni W, Song C, Li K, Gurzadyan GG, Guo X (2019) Defects promote ultrafast charge separation in graphitic carbon nitride for enhanced visible-light-driven CO2 reduction activity. Chem Eur J 25:5028–5035. https://doi.org/10.1002/chem.201805923
Li F, Yue X, Zhang D, Fan J, **ang Q (2021) Targeted regulation of exciton dissociation in graphitic carbon nitride by vacancy modification for efficient photocatalytic CO2 reduction. Appl Catal B 292:120179. https://doi.org/10.1016/j.apcatb.2021.120179
Yang Z, Zhang Y, Zhang H, Zhao J, Shi H, Zhang M, Yang H, Zheng Z, Yang P (2022) Nitrogen vacancies in polymeric carbon nitrides promote CO2 photoreduction. J Catal 409:12–23. https://doi.org/10.1016/j.jcat.2022.03.016
Yang P, Shang L, Zhao J, Zhang M, Shi H, Zhang H, Yang H (2021) Selectively constructing nitrogen vacancy in carbon nitrides for efficient syngas production with visible light. Appl Catal B 297:120496. https://doi.org/10.1016/j.apcatb.2021.120496
Hou J, Yang M, Dou Q, Chen Q, Wang X, Hu C, Paul R (2022) Defect engineering in polymeric carbon nitride with accordion structure for efficient photocatalytic CO2 reduction and H2 production. Chem Eng J 450:138425. https://doi.org/10.1016/j.cej.2022.138425
Wang R, Wang Z, Wan S, Ding J, Zhong Q (2022) Enhanced light-driven CO2 reduction on metal-free rich terminal oxygen-defects carbon nitride nanosheets. J Colloid Interface Sci 608:2505–2514. https://doi.org/10.1016/j.jcis.2021.10.169
Song X, Zhang X, Li X, Che H, Huo P, Ma C, Yan Y, Yang G (2020) Enhanced light utilization efficiency and fast charge transfer for excellent CO2 photoreduction activity by constructing defect structures in carbon nitride. J Colloid Interface Sci 578:574–583. https://doi.org/10.1016/j.jcis.2020.06.035
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871. https://doi.org/10.1103/PhysRev.136.B864
Wang Y, Tian Y, Yan L, Su Z (2018) DFT study on sulfur-doped g-C3N4 nanosheets as a photocatalyst for CO2 reduction reaction. J Phys Chem C 122:7712–7719. https://doi.org/10.1021/acs.jpcc.8b00098
Gao G, Jiao Y, Waclawik ER, Du A (2016) Single atom (Pd/Pt) supported on graphitic carbon nitride as an efficient photocatalyst for visible-light reduction of carbon dioxide. J Am Chem Soc 138:6292–6297. https://doi.org/10.1021/jacs.6b02692
Fu J, Liu K, Jiang K, Li H, An P, Li W, Zhang N, Li H, Xu X, Zhou H, Tang D, Wang X, Qiu X, Liu M (2019) Graphitic carbon nitride with dopant induced charge localization for enhanced photoreduction of CO2 to CH4. Adv Sci 6:1900796. https://doi.org/10.1002/advs.201900796
Wan S, Ou M, Wang X, Wang Y, Zeng Y, Ding J, Zhang S, Zhong Q (2019) Facile fabrication of oxygen and carbon co-doped carbon nitride nanosheets for efficient visible light photocatalytic H2 evolution and CO2 reduction. Dalton Trans 48:12070–12079. https://doi.org/10.1039/C9DT02507C
Wang X, Li Q, Gan L, Ji X, Chen F, Peng X, Zhang R (2021) 3D macropore carbon-vacancy g-C3N4 constructed using polymethylmethacrylate spheres for enhanced photocatalytic H2 evolution and CO2 reduction. J Energy Chem 53:139–146. https://doi.org/10.1016/j.jechem.2020.05.001
Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korean government (MSIT) (2022R1A2C3004242), and partly by the Ministry of Trade, Industry Energy (MOTIE) of the Republic of Korea under Grant No. 20019175.
Author information
Authors and Affiliations
Contributions
Pradeepta Babu (PB) conceptualized the review content and drafted the original version. Hyewon Park (HP) aided Pradeepta Babu (PB) in creating figures and tables. Jeong Young Park (JYP) provided overall supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Jeong Young Park is a member of the editorial board of this journal. He was not involved in the editorial review or the decision to publish this article. All authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Babu, P., Park, H. & Park, J.Y. Surface chemistry of graphitic carbon nitride: do** and plasmonic effect, and photocatalytic applications. Surf. Sci. Tech. 1, 29 (2023). https://doi.org/10.1007/s44251-023-00026-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44251-023-00026-1