
Abstract
Background and Objective
Alemtuzumab (Campath®) is used to prevent graft-versus-host disease and graft failure following pediatric allogeneic hematopoietic cell transplantation. The main toxicity includes delayed immune reconstitution, subsequent viral reactivations, and leukemia relapse. Exposure to alemtuzumab is highly variable upon empirical milligram/kilogram dosing.
Methods
A population pharmacokinetic (PK) model for alemtuzumab was developed based on a total of 1146 concentration samples from 206 patients, aged 0.2–19 years, receiving a cumulative intravenous dose of 0.2–1.5 mg/kg, and treated between 2003 and 2015 in two centers.
Results
Alemtuzumab PK were best described using a two-compartment model with a parallel saturable and linear elimination pathway. The linear clearance pathway, central volume of distribution, and intercompartmental distribution increased with body weight. Blood lymphocyte counts, a potential substrate for alemtuzumab, did not impact clearance.
Conclusion
The current practice with uniform milligram/kilogram doses leads to highly variable exposures in children due to the non-linear relationship between body weight and alemtuzumab PK. This model may be used for individualized dosing of alemtuzumab.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Alemtuzumab pharmacokinetics (PK) can be predicted using a population PK model, being the first step towards an individualized dosing regimen. |
Body weight is the most important covariate predicting PK. |
Blood lymphocyte counts, being a potential substrate for alemtuzumab, do not impact clearance. |
1 Introduction
Allogeneic hematopoietic cell transplantation (HCT) is a potentially curative treatment option for children with a variety of underlying diseases, including malignancies and benign disorders. Approaches to reduce mortality are essential, including the prevention of graft-versus-host disease (GvHD), which contributes to morbidity and mortality following HCT [1, 2].
Alemtuzumab (Campath®), a humanized anti-CD52 monoclonal antibody, was introduced as serotherapy to prevent GvHD and graft failure by in vivo depletion of lymphocytes [3]. The inclusion of alemtuzumab in the conditioning regimen significantly reduces the incidence of both acute and chronic GvHD [4,5,6]. An exposure-dependent relationship between alemtuzumab concentrations and acute GvHD was reported [1]. Conversely, higher doses of alemtuzumab have been associated with delayed immune reconstitution (IR) by excessive lymphodepletion [7, 8]. IR, especially of T cells, is dependent on peripheral expansion of graft-infused cells during the first months after HCT; depletion of these T cells may leave patients with no or little IR [9], which could potentially lead to increased viral reactivations as well as less graft-versus-leukemia effect, thereby abrogating the beneficial effect of GvHD reduction on overall survival (OS). Despite a reduced incidence of GvHD, the absence of improvement in OS with the inclusion of alemtuzumab may be due to delayed T-cell IR [1, 4, 5, 8, 10,11,12]. Moreover, most studies report on alemtuzumab pharmacology in adult populations; few studies have been performed in pediatric populations.
While evidence suggests a relationship between the use of alemtuzumab and clinical outcomes in adult populations, individual exposure of alemtuzumab is unpredictable due to highly variable pharmacokinetics (PK) [13,14,15,16] with the currently applied fixed empirical dosing in adults. As a consequence, patients treated with a comparable dose of alemtuzumab may have significant differences in drug exposure and, consequently, clinical outcome. Mostly descriptive PK of alemtuzumab are available in pediatric populations [17, 18], while variability in PK is often most substantial in children [19, 20]. The variable PK and their potential associations with outcome underline the need for predictable exposure to antibodies in all patients [13, 21,22,23,24]. Accordingly, the importance of dose individualization and/or therapeutic drug monitoring (TDM) of monoclonal antibodies is increasingly recognized [25, 26].
There is a great need for a population PK model for alemtuzumab in children in order to understand the PK. In future, dosing may be amended based on this PK model. In this study, we describe the population PK of alemtuzumab in children receiving an HCT as a first step to develo** an individualized dosing regimen.
2 Methods
2.1 Study Design and Patients
Patients receiving an HCT with alemtuzumab as part of their conditioning, and treated at the pediatric wards of the Leiden University Medical Center (LUMC), Leiden, The Netherlands, and Great Ormond Street Hospital (GOSH), London, UK, between January 2003 and July 2015, were included. In case of multiple HCTs per patient, all transplantations were included. Patients using other serotherapy drugs (antithymocyte globulin; ATG) within the same conditioning regimen were excluded, including patients who received alemtuzumab following allergic reactions to ATG in the same conditioning. Additionally, patients who received any type of serotherapy in a 3-month period before this HCT were excluded from this analysis. No restrictions were applied on the timing and dose of alemtuzumab, or any patient-, disease- or transplantation-related factors. Data were collected and samples were taken after informed consent was given through the parents and/or the child in accordance with the Declaration of Helsinki. Ethical committee approval was acquired through trial numbers P01.028 (Leiden) and V0904 (London).
Alemtuzumab (Campath®; Genzyme, Cambridge, MA, USA) was administered as an intravenous infusion, usually 6–8 days before HCT, for 4–5 consecutive days. In London, alemtuzumab was the standard choice for serotherapy, while in Leiden, alemtuzumab was reserved for patients with selected immune deficiencies and myelodysplastic syndrome. Patients with hemophagocytic lymphohistiocytosis (HLH) received alemtuzumab 15 days before transplantation. Although the dose of alemtuzumab varied, most patients were administered a cumulative dose of 1 mg/kg (5 × 0.2 mg/kg/day), with a substantial number of patients receiving alemtuzumab at a cumulative dose of 0.5 mg/kg at the treating physician’s discretion. Patients received antihistamines and high-dose corticosteroids around alemtuzumab infusions, in accordance with institutional guidelines. A small number of patients received in vitro lymphodepletion of the graft by direct addition of alemtuzumab 20 mg to the graft infusion bag 30 min prior to infusion [27]. Due to the small time window compared with the long half-life between adding alemtuzumab to the infusion bag containing the graft and the graft infusion itself, the full amount of alemtuzumab was assumed to be administered with infusion of the graft in these patients.
Conditioning regimens were administered according to (inter)national protocols (European Society for Bone Marrow Transplantation, Working Parties’ recommendations). GvHD prophylaxis consisted of cyclosporin A (controlled with TDM at trough levels of 150–250 μg/L) combined with either prednisolone (cord blood transplants) or methotrexate (matched unrelated donor). Patients receiving an identical related donor transplantation or CD34 selected graft did not receive any additional GvHD prophylaxis. All patients received gut decontamination and were treated in positive pressure, particle free, air-filtered, isolation rooms.
Samples for PK measurements were taken before and after each infusion, followed by one sample weekly in patients from Leiden, until approximately + 70 days after HCT, while, in London, three samples were available per patient—on the day of HCT (day 0), and + 14 and + 28 days after HCT. The samples around infusion were taken at ± 15 min before and after infusion, which, given the long half-life of alemtuzumab, can be seen as true trough and peak levels. Samples were prospectively collected and measured in batches.
2.2 Measurement of Alemtuzumab Concentration and Anti-Alemtuzumab Antibodies
2.2.1 Quantitative Flow Cytometry (Q-FACS) Assay
The laboratory in London, measuring samples from part of the London population, used a quantitative flow cytometry (Q-FACS) assay. Alemtuzumab levels were measured using Q-FACS assays, in modifications of the method described [28]. In short, 1 × 106 human T-cell line-78 (HUT-78) T cells were incubated using fourfold dilutions of patients’ serum in phosphate-buffered saline (PBS), followed by washing and incubation with conjugated secondary antibodies (Alexa Fluor 647 labeled goat anti-human IgG; Life Technologies). Incubations were carried out at room temperature for 30 min each. To construct a reference curve, HUT cells were incubated with known amounts of alemtuzumab (range 10–0.01 µg/mL) containing 25% human serum. Cells were washed and mean fluorescence intensity (MFI) was measured on an FACS Calibur machine (Becton Dickinson Biosciences, Franklin Lakes, NJ, USA). The lower limit of quantitation for alemtuzumab in this assay was 0.1 µg/mL, with a linear response from 0.01 to 0.1 μg/mL. The assay did not change over time.
2.2.2 Enzyme-Linked Immunosorbent Assay
All samples from Leiden, and part of the samples from London, were measured in Leiden using an enzyme-linked immunosorbent-based assay (ELISA) [29]. Microtiter plates (Corning Corporation, Corning, NY, USA) were coated with a human polyclonal anti-idiotype antibody to alemtuzumab (Geoff Hale Developments, Marston, Oxford, UK), diluted in PBS at a concentration of 0.5 µg/mL, by incubating overnight at 4 °C, followed by blocking with 2% casein in PBS. Samples, controls, and a diluted standard range of alemtuzumab (25.000–100 µg/mL, diluted in 10% pooled human serum) were applied and incubated at 37 °C for 1 h at room temperature. After washing, bound alemtuzumab was detected with biotin-labeled anti-idiotype antibody, 1 h at room temperature, followed by streptavidin poly-horseradish peroxidase (HRP; Sanquin, 8000145253, 2 µg/mL), for 30 min. The lower limit of detection was 0.01 µg/mL. The assay did not change over time.
In both assays, alemtuzumab spiked sera were used as controls. The results of 146 samples tested with both ELISA NC anti-idiotype and Q-FACS were compared. For the correlation, only samples with a measured alemtuzumab concentration > 0.1 µg/mL in Q-FACS were used. Electronic supplementary Fig. S1 shows the reasonable correlation of both assays (R2 = 0.89).
2.2.3 Population Pharmacokinetic (PK) Analysis
For analysis of the PK data, non-linear mixed-effects modeling was employed using NONMEM 7.3.0 (Icon Development Solutions LLC, Hanover, MD, USA). R version 3.2.3 and Pirana version 2.8.2 were used for preparation and visualization of data. First-order conditional estimation (FOCE) with interaction was used throughout the model development. Alemtuzumab concentrations were logarithmically transformed and simultaneously fitted. Samples that were reported to be below the limit of quantification (BLQ), which only occurred in the tail-end of the concentration, were set at half the BLQ, with subsequent samples being removed in accordance with method M6 [30]. Interindividual variability on PK parameters was assumed to follow a log-normal distribution, and were implemented in the model according to Eq. 1:
where \(P_{i}\) is the individual or post hoc value of the parameter in the ith individual, \(P_{\text{pop}}\)is the population mean for this parameter, and \(\eta_{i}\) is the interindividual variability of the ith person, which samples from a normal distribution with a mean of 0 and a variance of ω2. An additive error model was used, which, due to logarithmically transformed data, should be seen as a proportional error model. Here, the jth observation for the ith individual was described using Eq. 2:
where \(Y_{i,j}\) is the observed concentration, \(C_{{{\text{pred}},i,j}}\) is the jth predicted concentration for individual i, and \({\varepsilon }\)is the error, sampled from a normal distribution with a mean of 0 and a variance of σ2.
Several criteria were applied in the process of model building and selection. A decrease in objective function value (OFV) over 3.84 points between nested models was considered statistically significant; this correlated with p < 0.05 based on a Chi-square distribution with 1 degree of freedom. Goodness-of-fit (GOF) plots were evaluated, including observed versus both individual- and population-predicted concentrations, as well as conditional weighted residuals (CWRES) versus time and observed concentrations. Additionally, parameter uncertainty and \(\eta\)shrinkage were evaluated to assess model performance.
Interoccasion variability (IOV) was tested to assess changes in parameters between the respective doses according to Eq. 3:
where, compared with Eq. 1, κi is the IOV for the mth occasion. Individual PK parameters (post hocs) were estimated using the POSTHOC option in NONMEM.
The elimination of antibodies is often dependent on the concentration of substrate [31, 32], therefore non-linear elimination pathways were explored. No data on target concentrations over time (i.e. CD52 or lymphocytes) were available, therefore full target-mediated drug disposition (TMDD) models, as previously described, were not persued [32, 33]. Instead, non-linear elimination pathways were explored by incorporating clearance (CL) described by Michaelis–Menten kinetics (Eq. 4):
where V is the elimination rate, Vmax is the maximum elimination rate, C is the alemtuzumab concentration, and Km is the Michaelis–Menten constant, the concentration at which 50% of the maximum elimination rate is reached.
2.2.4 Covariate Model
Patient characteristics, including body size parameters (actual body weight, age, body surface area), and transplant- and disease-specific variables (sex, underlying disease, stem cell source, number of HCTs received, treatment center) were studied as possible covariates for their relation with PK parameters. In line with previous reports, the role of lymphocyte counts on alemtuzumab PK was also investigated as CD52 is almost exclusively expressed on these cells. Cell counts drawn before the first infusion of alemtuzumab were available; the lymphocyte counts are greatly reduced after the first dose and were therefore not taken in the included patients. Therefore, we considered baseline lymphocyte counts drawn within 48 h before infusion of the first alemtuzumab dose as a covariate. No other biochemical markers were evaluated as a covariate.
To assess the covariate relations, post hocs, interindividual variability, and CWRES were plotted against covariates, both before and after inclusion of the covariates, to evaluate potential relationships. Stepwise covariate modeling was performed in parallel. Lastly, only those covariates where a physiological or pharmacological mechanism could be hypothesized were included. Continuous covariates such as age and body weight were tested in a linear and power function (Eqs. 5 and 6):
where Pi and Covi are the parameter and covariate value for the ith individual, respectively, \(P_{\text{pop}}\) is the population mean for this parameter, and Covmedian is the standardized value for the covariate. In the linear relationship equation (Eq. 5), l represents the slope factor of the linear function, while, in the power relationship equation (Eq. 6), k is the scaling factor. Additionally, because the influence of bodyweight on CL appeared more complex, variations of Eq. 6 were explored, where k is dependent on the covariate value of the ith individual, as proposed by Wang et al. [34]. and implemented in several other models [35, 36]. Evaluated variations included a maximum effective concentration (Emax) approach and a power function according to Eq. 7:
where k is the exponential scaling factor in Eq. 6, k0 is the value for the exponent for an individual with a hypothetical bodyweight of 0 kg, kmax is the maximum decrease of the exponent, k50 is the bodyweight at which 50% of kmax is reached, and ϒ is the Hill coefficient determining the steepness of the sigmoidal decline. In the power function, a represents the coefficient and b is the exponent. This model was developed in order to reflect the changing influence of weight with age during growth of the child.
Potential covariates were evaluated using forward inclusion and backward elimination, with a significance level of < 0.005 (− 7.9 points in OFV) and < 0.001 (− 10.8 points in OFV), respectively. Building of the covariate model was comparable with the development of the structural model. In addition, after inclusion of a covariate, a decline in unexplained interindividual variability had to be achieved before inclusion into the final model [37].
2.2.5 Model Evaluation
The model was thoroughly evaluated for robustness. To assess the predictive performance of the model, bootstrap analyses were performed, stratified on treatment center. One thousand datasets were created using random selection from the original dataset; the final model was fit to each data set. For each parameter, median values from the thousand fits for each parameter, as well as 95% confidence intervals (CIs), were compared with parameter estimates of the final model.
In addition, a normalized prediction distribution of errors (NPDE) was performed, where the prediction discrepancies are simulated, taking into account the correlation between observations in the same individual and the predictive distribution [38]. Finally, prediction-corrected visual predictive checks (VPCs) were created to assess the predictive performance of the final model compared with the measured concentrations.
3 Results
3.1 Patients
A total of 206 patients receiving 212 HCTs were included from the two treatment centers (Table 1). Median age was 4.8 years (range 0.2–19; 28 infants < 1 year) and median body weight was 17.2 kg. Fifty-four percent of patients received a cumulative alemtuzumab dose of 0.9–1.1 mg/kg, while 35% received a cumulative dose of < 0.9 mg/kg. The first dose was a median of 8 days before the graft infusion, ranging from 0 days (alemtuzumab added the transplant itself) to 21 days before transplantation. Most patients (52%) received an HCT to treat an immune deficiency; the most frequently used stem cell source was bone marrow. A total number of 1146 concentration samples were available for this analysis (median 5.4 samples per patient) (Fig. 1). A total of 180 samples (112 from Leiden patients, 68 from London patients, 14% of all samples) were BLQ; 76 were set as 0.05 μg/mL, 104 samples were excluded. The majority of the samples (84%, collected in 136 patients) were measured in Leiden. The dataset was pooled in order to include all patients treated in the treatment period. No optimal sampling design was incorporated. In patients from Leiden, peak-trough and washout samples were available. For the patients in London, samples were available on the day of stem cell infusion (day 0; 8 days after the first infusion of alemtuzumab), day + 14 and day + 28.

Concentration-time plots of all patients from LUMC (open circles) and GOSH (dots) on a a normal scale and b a log scale. Dashed line represents the Michaelis–Menten constant Km. The start of the first alemtuzumab treatment is defined as T = 0. LUMC Leiden University Medical Center, GOSH Great Ormond Street Hospital
3.2 Structural PK Model
A two-compartment model best described the PK of alemtuzumab (Table 2, Fig. 2, and electronic supplementary Fig. S7). Compared with a one-compartment model, the two-compartment model was superior in terms of GOF plots and OFV (253 points decrease in OFV; p < 0.001). However, large residual standard errors in the parameters associated with distribution were observed, as well as a high dependency on initial values. To address this issue, model simplification was applied, with peripheral volume of distribution (V2) being estimated as a factor of the central volume of distribution (V1), as previously shown in the literature [39,40,41], which made the model more stable and independent on initial values. This model yielded a decrease of 158 points in OFV compared with the one-compartment model (p < 0.001), and showed comparable GOF plots compared with the full two-compartment model. A three-compartment model proved unstable, showing inaccurate parameter estimates. A proportional error model was incorporated in the model.

Goodness-of-fit plots of the final model: individual predicted versus observed concentrations of alemtuzumab in all patients, split by quartiles of body weight. a < 11 kg; b 11–17.3 kg; c 17.3–32 kg; and d > 32 kg. Lines represent the line of unity (x = y)
Looking at the individual concentration-time profiles, non-linear PK features are suggested by the bell-shaped curve at higher concentrations (Fig. 1b). Models with only non-linear CL, as well as models with parallel linear and non-linear CL, were therefore evaluated. In this study, compared with only linear CL, both models resulted in a substantial decrease in OFV, with the model with parallel CL pathways being clearly superior (− 39 and − 99 points in OFV for only non-linear and parallel CL with three and four additional parameters, respectively). Therefore, alemtuzumab elimination was described using linear and non-linear CL, which was parameterized using the Michaelis–Menten equation incorporating the maximum elimination rate (Vmax) and Michaelis–Menten constant (Km), depicting the concentration at which the elimination rate was 50% of the Vmax. Besides a decrease of 99 points in OFV, the addition of non-linear CL to the linear CL model resulted in an improvement in GOF plots. The Michaelis–Menten constant could be well-estimated and fell within the observed concentration range (Fig. 1). The relative contribution of linear and non-linear CL is depicted in electronic supplementary Fig. S2. No improvement of the model in terms of OFV and GOF plots was observed when including IOV on any of the parameters.
3.3 Covariate Model
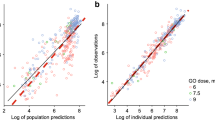
According to the predefined criteria, the covariate analysis showed that actual body weight and age were correlated with central volume of distribution and linear CL. Actual body weight proved the best predictor for these parameters, both in terms of decrease in OFV and improvement of GOF plots (Fig. 3). The inclusion of body weight as a power function (Eq. 6) on V1 yielded a decrease in OFV of 92 points. In addition, the effect of body weight on linear CL was parameterized as a body-weight-dependent exponent (BDE), in which the exponent differs according to body weight (Eq. 7, Fig. 3) [35, 36]. Including a BDE parameterization on linear CL gave a better description of the relation with body weight, especially in smaller children, as seen in plots of interindividual variability on CL versus body weight, and gave a 52-point decline in OFV. The exponent in this model varied from 3.69 in children with a body weight of 5 kg to 0.41 in patients weighing 80 kg (electronic supplementary Fig. S3). Finally, body weight was introduced as a covariate on intercompartmental distribution based on the described relationship in the literature. This yielded another decrease in OFV of 22 points, and gave a substantial improvement in GOF plots split for age (Fig. 2).

Interindividual variability on clearance (upper panels) and central volume of distribution (lower panels), both before (left plots) and after (right plots) the inclusion of body weight
Baseline lymphocyte counts were evaluated as a covariate for CL. Data on baseline lymphocyte counts were missing in 56 patients; these were set at median baseline lymphocyte count in the population. Baseline lymphocyte counts did not influence any PK parameters, including linear and non-linear CL (electronic supplementary Figs. S4 and S5). Even when removing patients with missing values for baseline lymphocytes from the dataset, no effect of baseline lymphocytes on CL were found. The full model code can be found in the electronic supplementary data.
3.4 Internal Validation
The final model with body weight on volume of distribution, intercompartmental distribution, and in a BDE parameterization on linear CL was stable in bootstrap analysis (96.1% successful). The bootstrap was stratified on treatment center to account for the density of sampling. Median and 95% CIs were in line with the model estimations and residual standard errors (Table 2). The NPDE analysis showed normally distributed errors, with no major trends in NPDE versus time or NPDE versus predictions. The prediction-corrected VPC shows model simulations to be in line with model predictions, both in high and low concentrations (Fig. 4).

Evaluation studies. a–c NPDE. a Histogram of the NPDE, with the solid line representing a normal distribution with a mean of 0 and variance of 1. b NPDE versus time. c NPDE versus predictions. Grey blocks represent the 95% CI of the NPDE. Prediction-corrected VPC on d a normal axis and e a logarithmically transformed axis. Solid lines represent the 5% CIs, median and 95% CI of the data; dotted lines represent the 5% CI, median and 95% CI of the simulations; dark grey blocks represent the median of the simulations; and light grey blocks represent the 95% CIs of the simulations. NDPE normalized prediction distribution of errors, CI confidence interval, VPC visual predictive check
4 Discussion
Alemtuzumab plays an important role in preventing GvHD and graft failure following pediatric HCT, as well as the occurrence of early T-cell IR [1, 2, 6, 42]. In this large cohort of children, we describe the population PK of alemtuzumab in an HCT setting. The proposed model adequately describes the observed concentrations, and was extensively validated. Actual body weight was found to be a predictor for CL and central volume of distribution, and should therefore be taken into account for dosing of alemtuzumab.
In the developed model, alemtuzumab elimination was best described using a parallel linear and saturable CL pathway. This is in line with antibody pharmacology, where both target binding and non-specific degradation are the major elimination pathways [43, 44]. The implemented parameterization with parallel linear and non-linear CL pathways, the latter using Michaelis–Menten kinetics, is often used, particularly when the antibody targets a non-soluble protein [31]. As lymphocytes harbor the vast majority of CD52 [45] the peripheral blood baseline lymphocyte count was considered as a covariate for elimination. However, no impact of baseline lymphocyte counts on any PK parameters was found. A possible explanation could be that an excess of drug is introduced in relation to the amount of CD52, thereby minimizing the effect of target availability. However, this should be kept in mind when significantly decreasing the administered dose as cell counts are known to influence alemtuzumab CL at lower dosages [13].
A previous study by Mould et al. described the population PK of alemtuzumab in adults treated for chronic lymphatic leukemia (CLL) [13]. Alemtuzumab PK were described using a two-compartment model incorporating saturable CL, no linear CL was included in the model. White blood cell (WBC) count on Vmax was found to be the only covariate predicting PK, indicating a higher maximal CL rate in patients harboring more targets for alemtuzumab. Although the population and treatment setting in the current study is significantly different, our parameter estimates in terms of total CL and central volume of distribution are broadly similar to their results. Importantly, the dosage and usage of alemtuzumab in a CLL setting is markedly different from conditioning in HCT. Where alemtuzumab is used long-term for tumor suppression in CLL, its use in HCT is rapid and results in full depletion of recipient T cells. This is reflected in the substantially higher dose in HCT, which leads to full depletion of lymphocytes over the course of 2 days. This may explain why the CLL study did indeed find that cell counts impacted the elimination of alemtuzumab, while, in an HCT setting using high doses of alemtuzumab, the role of cell counts on the PK is minor. Another population PK study including 17 children, presented as conference proceeding, described alemtuzumab PK in pediatric HCT using a one-compartment model, with only weight as a covariate on CL and volume [18]. No relationship between lymphocytes and PK parameters was identified in this study.
Few studies have investigated the dose–effect or exposure–effect relationship of alemtuzumab in terms of IR. Nonetheless, T-cell reconstitution, especially of CD3+ and CD4+ T cells, is suggested to be slower following higher exposures of alemtuzumab [1, 8, 46]. In terms of clinical outcome parameters, higher doses of alemtuzumab have been associated with a lower incidence of GvHD [1, 4, 5, 10, 12, 47, 48]. In one study investigating alemtuzumab concentration rather than dosage, those patients with higher concentrations on the day of HCT had less acute GvHD, but more mixed chimerism and poor IR; however, no impact on survival was demonstrated based on the concentrations on the day of HCT [1].
There are some weaknesses in this study. Foremost, the study was not designed for population PK analysis, and therefore the timing of sample collection was not optimized. As a consequence, approximately only one-third of patients had peak/trough concentrations available, making the estimation of volumes of distribution difficult. This is reflected in a minor underestimation of peak concentrations. Second, samples were measured in two laboratories using different assays. Although validation studies show an adequate correlation between both assays, and laboratory was not a covariate in the model, the model might have been more stable when all samples were measured in the same laboratory. Furthermore, the unexplained variability on CL and Km remains substantial. This makes it more difficult to give dose recommendations for future patients, and TDM may be needed to correct for unexplained variability in the PK. Next, interindividual variability is rather high and cannot be well explained by the introduction of the available covariates. This particularly applies to the high variability on CL of 104%. This limits the possibilities of the model to predict individual concentrations in future patients, and may suggest that TDM is needed.
Finally, the parameterization of the covariate effect of body weight on linear CL was optimized to describe changes across the entire weight range. However, data from only a few individuals (n =10) with a body weight higher than 60 kg were available. As a consequence, the predictive value above this weight range may be lower than for other parts of the curve. This applies particularly to the fact that linear CL is expected to slightly decrease from a body weight of 60 kg onwards (decrease of 1.2%).
5 Conclusions
We have developed and extensively validated a population PK model that adequately describes alemtuzumab PK over the entire pediatric age range. This model incorporates parallel linear and non-linear elimination pathways, reflecting TMDD as frequently observed in antibody kinetics. Actual body weight was identified as a covariate on CL, volume of distribution, and intercompartmental distribution, the former as a BDE. Although CD52 is mainly expressed on lymphocytes, no relationship between baseline lymphocyte counts and alemtuzumab elimination was found. Evaluation of the current dosing regimen showed that exposure varies across age and is therefore suboptimal.
This model can be used for further studies to investigate optimal alemtuzumab exposure. Once the therapeutic window is identified, the model may serve as the basis for an individualized dosing regimen for children receiving an HCT. Using this regimen, optimal alemtuzumab exposure can be achieved, potentially improving clinical outcome in these children.
References
Marsh RA, Lane A, Mehta PA, et al. Alemtuzumab levels impact acute GVHD, mixed chimerism, and lymphocyte recovery following alemtuzumab, fludarabine, and melphalan RIC HCT. Blood. 2015;127(4):503–13.
Kanda J, Lopez RD, Rizzieri DA. Alemtuzumab for the prevention and treatment of graft-versus-host disease. Int J Hematol. 2011;93(5):586–93.
Kottaridis PD, Milligan DW, Chopra R, et al. In vivo CAMPATH-1H prevents graft-versus-host disease following nonmyeloablative stem cell transplantation. Blood. 2000;96(7):2419–25.
Perez-Simon JA, Kottaridis PD, Martino R, et al. Nonmyeloablative transplantation with or without alemtuzumab: Comparison between 2 prospective studies in patients with lymphoproliferative disorders. Blood. 2002;100(9):3121–7.
van Besien K, Kunavakkam R, Rondon G, et al. Fludarabine-melphalan conditioning for AML and MDS: alemtuzumab reduces acute and chronic GVHD without affecting long-term outcomes. Biol Blood Marrow Transplant. 2009;15(5):610–7.
Poire X, van Besien K. Alemtuzumab in allogeneic hematopoetic stem cell transplantation. Expert Opin Biol Ther. 2011;11(8):1099–111.
Spyridonidis A, Liga M, Triantafyllou E, et al. Pharmacokinetics and clinical activity of very low-dose alemtuzumab in transplantation for acute leukemia. Bone Marrow Transplant. 2011;46(10):1363–8.
Lane JP, Evans PT, Nademi Z, et al. Low-dose serotherapy improves early immune reconstitution after cord blood transplantation for primary immunodeficiencies. Biol Blood Marrow Transpl. 2014;20(2):243–9.
Booth C, Veys P. T cell depletion in paediatric stem cell transplantation. Clin Exp Immunol. 2013;172(2):139–47.
Chakraverty R, Orti G. Impact of in vivo alemtuzumab dose before reduced intensity conditioning and HLA-identical sibling stem cell transplantation: pharmacokinetics, GVHD, and immune reconstitution. Blood. 2010;116(16):3080–8.
Cook G, Smith GM, Kirkland K, et al. Outcome following reduced-intensity allogeneic stem cell transplantation (RIC AlloSCT) for relapsed and refractory mantle cell lymphoma (MCL): a study of the British society for blood and marrow transplantation. Biol Blood Marrow Transplant. 2010;16(10):1419–27.
Malladi RK, Peniket AJ, Littlewood TJ, et al. Alemtuzumab markedly reduces chronic GVHD without affecting overall survival in reduced-intensity conditioning sibling allo-SCT for adults with AML. Bone Marrow Transpl. 2009;43(9):709–15.
Mould DR, Baumann A, Kuhlmann J, et al. Population pharmacokinetics-pharmacodynamics of alemtuzumab (Campath) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br J Clin Pharmacol. 2007;64(3):278–91.
Elter T, Molnar I, Kuhlmann J, Hallek M, Wendtner C. Pharmacokinetics of alemtuzumab and the relevance in clinical practice. Leuk Lymphoma. 2008;49(12):2256–62.
Rebello P, Cwynarski K, Varughese M, Eades A, Apperley JF, Hale G. Pharmacokinetics of CAMPATH-1H in BMT patients. Cytotherapy. 2001;3(4):261–7.
Morris EC, Rebello P, Thomson KJ, et al. Pharmacokinetics of alemtuzumab used for in vivo and in vitro T-cell depletion in allogeneic transplantations: relevance for early adoptive immunotherapy and infectious complications. Blood. 2003;102(1):404–6.
Marsh RA, Fukuda T, Emoto C, et al. Pretransplant absolute lymphocyte counts impact the pharmacokinetics of alemtuzumab. Biol Blood Marrow Transplant. 2017;23(4):635–41.
Fukuda T, Emoto C, Marsh R, Neumeier L, Vinks AA, Mehta PA. Precision dosing of alemtuzumab: Population pharmacokinetic modeling in pediatric patients undergoing allogeneic hematopoietic cell transplantation for non-malignant diseases. Blood. 2016;128(22):2203.
Admiraal R, van Kesteren C, Boelens JJ, Bredius RGM, Tibboel D, Knibbe CAJ. Towards evidence-based dosing regimens in children on the basis of population pharmacokinetic pharmacodynamic modelling. Arch Dis Child. 2014;99(3):267–72.
Knibbe CAJ, Danhof M. Individualized dosing regimens in children based on population PKPD modelling: are we ready for it? Int J Pharm. 2011;415(1–2):9–14.
Admiraal R, van Kesteren C, Jol-van Der Zijde CM, et al. Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haematopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol. 2015;2(5):e194–e203.
Yang J, Zhao H, Garnett C, et al. The combination of exposure-response and case-control analyses in regulatory decision making. J Clin Pharmacol. 2013;53(2):160–6.
Chiu Y-L, Rubin DT, Vermeire S, et al. Serum adalimumab concentration and clinical remission in patients with Crohn’s disease. Inflamm Bowel Dis. 2013;19(6):1112–22.
Wierda WG, Kipps TJ, Keating MJ, et al. Self-administered, subcutaneous alemtuzumab to treat residual disease in patients with chronic lymphocytic leukemia. Cancer. 2011;117(1):116–24.
Mould DR, D’Haens G, Upton RN. Clinical decision support tools: the evolution of a revolution. Clin Pharmacol Ther. 2016;66(5):732–40.
Oude Munnink T, Henstra M, Segerink L, Movig K, Brummelhuis-Visser P. Therapeutic drug monitoring of monoclonal antibodies in inflammatory and malignant disease—translating TNF-α experience to oncology. Clin Pharmacol Ther. 2015;99(4):1–23.
Von Dem Borne PA, Beaumont F, Starrenburg CWJI, et al. Outcomes after myeloablative unrelated donor stem cell transplantation using both in vitro and in vivo T-cell depletion with alemtuzumab. Haematologica. 2006;91(11):1559–62.
Rebello P, Hale G. Pharmacokinetics of CAMPATH-1H: assay development and validation. J Immunol Methods. 2002;260(1–2):285–302.
Jol-van der Zijde C, Bredius R, Jansen-Hoogendijk A, et al. IgG antibodies to ATG early after pediatric hematopoietic SCT increase the risk of acute GVHD. Bone Marrow Transplant. 2012;47(3):360–68.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504.
Yan X, Mager DE, Krzyzanski W. Selection between Michaelis–Menten and target-mediated drug disposition pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2010;37(1):25–47.
Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35(5):573–91.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
Wang C, Peeters M, Allegaert K, et al. A bodyweight-dependent allometric exponent for scaling clearance across the human life-span. Pharm Res. 2012;29(6):1570–81.
Bartelink IH, Boelens JJ, Bredius RGM, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012;51(5):331–45.
Ince I, De Wildt SN, Wang C, et al. A novel maturation function for clearance of the cytochrome P450 3A substrate midazolam from preterm neonates to adults. Clin Pharmacokinet. 2013;52(7):555–65.
Krekels EHJ, van Hasselt JGC, Tibboel D, Danhof M, Knibbe CAJ. Systematic evaluation of the descriptive and predictive performance of paediatric morphine population models. Pharm Res. 2011;28(4):797–811.
Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90(2):154–66.
Admiraal R, van Kesteren C, Jol-van der Zijde CM, et al. Population pharmacokinetic modeling of Thymoglobulin® in children receiving allogeneic-hematopoietic cell transplantation (HCT): towards improved survival through individualized dosing. Clin Pharmacokinet. 2015;54(4):435–46.
De Cock RFW, Allegaert K, Schreuder MF, et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet. 2012;51(2):105–17.
Knibbe CAJ, Krekels EHJ, van den Anker JN, et al. Morphine glucuronidation in preterm neonates, infants and children younger than 3 years. Clin Pharmacokinet. 2009;48(6):371–85.
Soiffer RJ, Lerademacher J, Ho V, et al. Impact of immune modulation with anti-T-cell antibodies on the outcome of reduced-intensity allogeneic hematopoietic stem cell transplantation for hematologic malignancies Impact of immune modulation with anti-T-cell antibodies on the outcome of reduc. Blood. 2011;117(25):6963–70.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507.
Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24(1):23–39.
Zhao Y, Su H, Shen X, Du J, Zhang X, Zhao Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm Res. 2017;66(7):571–8.
Willemsen L, Jol-van der Zijde CM, Admiraal R, et al. Impact of serotherapy on immune reconstitution and survival outcomes after stem cell transplantations in children: thymoglobulin versus alemtuzumab. Biol Blood Marrow Transplant. 2015;21(3):473–82.
Lane JP, Evans PTG, Nademi Z, et al. Low-dose serotherapy improves early immune reconstitution after cord blood transplantation for primary immunodeficiencies. Biol Blood Marrow Transplant. 2014;20(2):243–9.
Peggs KS, Sureda A, Qian W, et al. Reduced-intensity conditioning for allogeneic haematopoietic stem cell transplantation in relapsed and refractory Hodgkin lymphoma: impact of alemtuzumab and donor lymphocyte infusions on long-term outcomes. Br J Haematol. 2007;139(1):70–80.
Acknowledgements
The authors would like to thank A.M. Jansen-Hoogendijk for measuring the samples; the data managers for collecting the patient characteristics and outcome data; Michel Koudijs for performing exploratory analyses; and the medical and nursing staff from the LUMC and GOSH for implementing this study protocol.
Author information
Authors and Affiliations
Contributions
RA designed and conducted the research, analyzed the data, and wrote the paper; CMJZ, SA, AE, and MW analyzed the samples and data and wrote the paper; CK and CAJK conducted the research, analyzed the data, and wrote the paper; GH supplied the anti-alemtuzumab antibodies; AL and JJB supervised the project and wrote the paper; RGMB, PV and JMFS included patients and wrote the paper.
Corresponding author
Ethics declarations
Funding
This study was funded by the Netherlands Organisation for Health Research and Development (ZonMW; Grant Number 40-41500-98-11044). The work of CAJK is supported by the Innovational Research Incentives Scheme (Vidi grant, June 2013) of the Dutch Organization for Scientific Research (NWO). This work was performed independently of all funders.
Conflict of interest
Rick Admiraal, Cornelia M. Jol-van der Zijde, Juliana M. Furtado Silva, Catherijne A.J. Knibbe, Arjan C. Lankester, Jaap Jan Boelens, Goeff Hale, Aniekan Etuk, Melanie Wilson, Stuart Adams, Paul Veys, Charlotte van Kesteren and Robbert G.M. Bredius have no conflicts of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Admiraal, R., Jol-van der Zijde, C.M., Furtado Silva, J.M. et al. Population Pharmacokinetics of Alemtuzumab (Campath) in Pediatric Hematopoietic Cell Transplantation: Towards Individualized Dosing to Improve Outcome. Clin Pharmacokinet 58, 1609–1620 (2019). https://doi.org/10.1007/s40262-019-00782-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00782-0