
Abstract
For conservation genetic studies using non-invasively collected samples, genome-wide data may be hard to acquire. Until now, such studies have instead mostly relied on analyses of traditional genetic markers such as microsatellites (SSRs). Recently, high throughput genoty** of single nucleotide polymorphisms (SNPs) has become available, expanding the use of genomic methods to include non-model species of conservation concern. We have developed a 96-marker SNP array for use in applied conservation monitoring of the Scandinavian wolverine (Gulo gulo) population. By genoty** more than a thousand non-invasively collected samples, we were able to obtain precise estimates of different types of genoty** errors and sample dropout rates. The SNP panel significantly outperforms the SSR markers (and DBY intron markers for sexing) both in terms of precision in genoty**, sex assignment and individual identification, as well as in the proportion of samples successfully genotyped. Furthermore, SNP genoty** offers a simplified laboratory and analysis pipeline with fewer samples needed to be repeatedly genotyped in order to obtain reliable consensus data. In addition, we utilised a unique opportunity to successfully demonstrate the application of SNP genotype data for reconstructing pedigrees in wild populations, by validating the method with samples from wild individuals with known relatedness. By offering a simplified workflow with improved performance, we anticipate this methodology will facilitate the use of non-invasive samples to improve genetic management of many different types of populations that have previously been challenging to survey.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Management and conservation of wide-ranging, elusive species occurring at low densities is challenging due to their hard-to-survey nature. Consequently, there is a need to develop novel tools for assessing vital information such as population size, sex ratio and demographic parameters (McMahon et al. 2011; Mills 2013; Pereira et al. 2013). Non-invasive survey methods focusing on genetic data, are becoming increasingly utilised for this (Beja-Pereira et al. 2009; Bruford et al. 2017; Carroll et al. 2018; Ferreira et al. 2018), where potential DNA sources, such as hair, saliva, scats and urine, are collected without interfering with natural behaviours or compromising individual survival (Rodgers and Janečka 2013). Such genetic data can form the basis for estimates of population size, survival and reproductive success, and provide insights into genotypic or environmental variables contributing to individual fitness (Bérénos et al. 2014). It can also identify cases of inbreeding and be used to inform genetic rescue operations (Åkesson et al. 2016; Vilà et al. 2003), thus providing predictive power for management decisions and applied conservation biology (Johnson et al. 2010; Soulé 1985; Soulé and Mills 1992).
The main limitations for non-invasively collected genetic samples are that they often contain low-quality and quantity of DNA, limiting the use of high-throughput genomic analysis methods (Carroll et al. 2018; but see Khan et al. 2020), and high cost of sequencing large number of samples (Aylward et al. 2018; Förster et al. 2018; Snyder-Mackler et al. 2016). Consequently, many genetic studies have been restricted to more traditional low-throughput genetic markers such as microsatellites (SSRs), hampering the uptake of genomics applications in practical conservation projects, and creating the so called “conservation genomics gap” (Shafer et al. 2015). Recently, genoty** of Single Nucleotide Polymorphism (SNP) markers, that offer increased precision, repeatability and resolution compared to classic SSR markers, has been applied to such low-quality DNA (Helyar et al. 2011; Kraus et al. 2015; Norman and Spong 2015; Nussberger et al. 2014; von Thaden et al. 2020). SNP markers offer many advantages compared to SSRs, like low error rates and low levels of homoplasy (Miller et al. 2011; Morin et al. 2009). However, practical limitations in the form of restricted financial and technical resources often lead to a need to reduce the amount of SNP markers, while maximizing the genomic resolution to render manageable and cost effective methods for genetic monitoring of wildlife.
In addition to individual identification, population structure inference and sexing, multi-locus SNP panels offer the possibility to infer both effective population size and precise local population sizes as well as relatedness between individuals (Norman and Spong 2015; Spitzer et al. 2016). Importantly, the high genetic resolution available from SNP data is useful for pedigree reconstruction (including the identification of mother, father and offspring triads) (Anderson and Garza 2006; Norman and Spong 2015). However, there are many challenges involved in reconstructing pedigrees; such as incomplete sampling, unknown population sizes, overlap** generations with long reproductive lifespans and lack of information on individual age. Age data is especially important for determination of the directionality of inferred parent – offspring relationships (Wang and Santure 2009). The level of genetic variability and inbreeding in the population also affects the precision in genetic pedigree reconstruction. Consequently, there is a need for studies validating pedigrees reconstructed solely from genetic data, to assess the risk of falsely inferred relationships as well as the influence of factors such as the number of markers used.
The wolverine (Gulo gulo) is a good example of a species that is hard to survey; an elusive and territorial carnivore with large home ranges, occurring at low densities, often in remote and inaccessible terrain (Inman et al. 2012; Persson et al. 2010). As a consequence, wolverines have been considered for protection as a threatened species under the Endangered Species Act in (the contiguous) United States (USFWS 2013). In Europe, the majority of the wolverines occur in the Scandinavian population (Norway and northern part of Sweden, Chapron et al. 2014), where it is red-listed as “Vulnerable” in Sweden (Swedish species information centre 2015) and “Endangered” in Norway (Henriksen and Hilmo 2015). A main challenge for wolverine conservation in Scandinavia is depredation conflicts caused by wolverine predation on free-ranging, semi-domestic reindeer (Rangifer tarandus) owned by indigenous Sámi reindeer-herding communities in both countries (Hobbs et al. 2012; Mattisson et al. 2016; Persson et al. 2015) and free ranging domestic sheep (Ovis aries) in Norway (Mattisson et al. 2016). Consequently, the Scandinavian wolverine population is intensively monitored to be kept above national management goals for minimum population size, set to minimize conflicts while ensuring population viability (SEPA 2014).
Here we describe the development of a set of 96 SNPs to be used within the conservation monitoring program for wolverines in Scandinavia. The SNP identification is based on data from a whole genome sequencing effort of individuals from the same population (Ekblom et al. 2018). We specifically present the validation process, focusing on the following: (1) Estimation of rates of genoty** errors, marker and sample dropouts in non-invasively collected samples. (2) Assessment of the number of SNP markers needed for reliable individual identification. (3) Evaluation of the performance of the new SNP-set in relation the classic SSR method for identification and DBY intron markers for sexing. (4) Benchmarking SNP genotype data for pedigree reconstruction. For pedigree validation, we use samples from a long-term, individual-based field study. Consequently, this provides detailed information about social system and relatedness for known age individuals in a local population (Aronsson and Persson 2018; Hedmark et al. 2007; Persson et al. 2010), offering a unique opportunity with important baseline information for validation of new procedures based on DNA, and specifically for reconstruction of pedigrees. Our high-throughput SNP genoty** procedure is optimised to handle thousands of samples with efficient lab and analysis workflows. The framework presented here offers practical advice for DNA-based population monitoring and conservation management. In addition, we provide general guidance on when and how to apply SNP markers in conservation genetic projects relying on non-invasive sampling of DNA samples.
Materials and methods
Study population and sampling
The Scandinavian wolverine population has been subject to extensive monitoring for over two decades (Aronsson and Persson 2017; Brøseth et al. 2010). During most of this time, genetic analyses have formed a central part of the monitoring program (Bischof et al. 2016, 2020; Ekblom et al. 2018), together with snow-tracking and searches for natal den sites. For more information regarding the monitoring program, see Aronsson and Persson (2017). Non-invasive DNA samples (mainly from scats, but also hair, urine, blood and secretions) are routinely collected in the field during the monitoring period, from February to June. In addition to verifying species, sex and individual identity, genetic data have been utilised to learn about population structure (Walker et al. 2001), genetic diversity (Ekblom et al. 2018), relatedness among individuals (Hedmark and Ellegren 2007), and local density estimates through a sampling-resampling framework (Bischof et al. 2016). The current population estimate is 1035 (95% CrI: 985–1088) wolverines in Scandinavia (660 in Sweden and 375 in Norway; Flagstad et al. 2019). Generations are overlap** with an average generation time of 6 years (Nilsson 2013), and many individuals are repeatedly sampled both within and between seasons.
For this study we used a total of 2005 DNA samples from wolverines collected in Sweden Norway and Finland. A majority of the samples (N = 1836) were non-invasively collected from 2001 to 2017 (mainly scats and secretions). These samples are referred to as “monitoring samples” hereafter. We also used tissue samples from dead individuals (N = 85, collected 2014–2017) obtained from the National Veterinary Institute, where autopsies of all encountered dead wolverines are routinely conducted, and these samples are referred to as “tissue samples”. In addition, we used tissue samples from 84 individuals collected from known individuals within a long-term study of wolverine ecology in Northern Sweden (1994–2011, The "Sarek study"; e.g. Aronsson and Persson 2017, 2018; Persson 2005; Persson et al. 2009), these samples are referred to as “Sarek samples”. From these individuals we have access to detailed demographic and spatial field data, including parental relationships.
For monitoring samples collected from 2015–2017 (N = 1654), a small piece of material was dissolved in Buffer TL (VWR) and treated overnight with Proteinase K (20 mg/ml) at 55 °C before automated DNA extraction using the Maxwell® 16 MDx Instrument and the Maxwell® 16 Tissue DNA Purification Kit (Promega). Samples collected before 2015 (N = 182) were extracted using either the QIAamp DNA stool mini kit (GmbH, Hilden, Germany)(Hedmark and Ellegren 2007) or a Genemole DNA extraction robot (Mole Genetics, Lysaker, Norway) (Bischof et al. 2016). Each extraction run included a negative control to detect cross contamination of samples and contamination of reagents. DNA from tissue samples and Sarek samples were extracted using the Qiagen DNeasy Blood & Tissue Kit. All monitoring and tissue samples had previously been genotyped and sexed using 19 SSRs (see Hedmark et al. 2004 for more information about the SSR markers and genoty** procedure).
Development of SNP marker-set
In a previous study of Scandinavian wolverines, thousands of high-quality and high information content SNPs were identified from the wolverine genome using re-sequencing, and several hundred of these were verified using independent genoty** (Ekblom et al. 2018). Furthermore, Ekblom et al. (2018) described a 96 SNP set suitable for genoty** of low-quality samples (hereafter referred to as “set A”). However, set A include markers with limited genoty** success and information content, and does not contain markers suitable for sexing. Consequently, we evaluated an additional SNP set (“set B”) with 65 autosomal SNP markers (not included in set A), 17 putative X- and 14 putative Y-chromosome markers. After preliminary analyses of both set A and B, a combined set of the best working markers was chosen to produce the final 96 SNP marker set selected for wolverine scat genoty** (“set D”). This included 87 autosomal SNPs, 6 X-chromosome linked SNPs and 3 Y-chromosome markers. Details and flanking sequences for all SNP markers in set A, B and D are available in Appendix S1. Y-chromosome markers are monomorphic, but only produce a genotype signal in samples from males, consequently these were only used for sexing and removed from the data for all other analyses. A combination of X-chromosome genotype and signal data from all 3 Y-chromosome markers was used for sex-identification (for details, see section “Sexing and individual identification”). All Sarek samples were also genotyped for the complete 375 SNP marker set from Ekblom et al. (2018) using the GoldenGate platform (Illumina). This set completely overlaps with all autosomal markers included in SNP sets A, B and D.
SNP genoty**
SNPs were genotyped using Fluidigm integrated fluidic circuits (IFCs) with 96 samples and 96 markers for each plate (96.96 Dynamic Array™ IFC). Prior to genoty**, all samples were pre-amplified using a highly multiplexed PCR, called Specific Target Amplification (STA). Here all 96 marker sites were simultaneously amplified using STA primers. Tissue samples were diluted to 5 ng/µl before performing the STA reaction and then run together with the non-invasively collected samples. The PCR was run for 40 cycles at 60 °C annealing/elongation temperature, other details were according to the manufacturer’s protocol. The STA products were then diluted to 1:10 in DNA Suspension Buffer. Genoty** markers, pre-amplified samples and control line fluid were loaded onto the IFC according to the manufacturer’s instructions. The IFC Controller was used for priming and loading the chip. The genoty** reactions were run on the FC1 Cycler according to the manufacturer’s instructions and reagents. This included thermal mix at 70 °C for 30 min and 25 °C for 10 min, hot start activation at 95 °C for 5 min, annealing temperature touch-down for 5 PCR cycles from 64 °C to 60 °C, followed by 26 cycles at 60 °C (each with 45 s annealing steps). The IFCs were finally analysed using the EP1 Reader. All samples were run in duplicate, including both positive and negative controls on each chip. Genotype calls were made using the Fluidigm SNP Genoty** Analysis software (version 4.3.1) using SNP-type normalisation, K-Means clustering and an automatic confidence threshold of 85, followed by manual inspection and correction of genotype clusters. Genotypes were exported as CSV files and converted into PLINK format using R version 3.3.1 (R Core Team 2016). R-scripts used for data handling and subsequent analyses are available as supporting information (Appendix S2).
Genotype analysis
The sample consensus genotype of each marker was set to “missing” if one or both of the duplicates for that sample did not produce a reliable genotype or if the two duplicates had different genotypes for the marker in question. Samples with marker dropout exceeding 15% or genotype mismatch between duplicates exceeding 5% were run in an additional duplicate (“re-run”), in order to obtain more reliable genotype calls. Samples where both duplicates had a marker dropout rate above 85%, and re-run samples where the marker dropout remained above 62%, were classified as non-working samples and were discarded for error rate analyses and individual assignment. Consensus genotypes of re-run samples were scored as homozygous only if two or more of the runs yielded a homozygous genotype and the second allele was completely absent from all runs. Heterozygous genotypes of re-run samples were called if more than one run yielded a heterozygous genotype. In other cases, the genotype of the marker was set to “missing”. All genotypes were also checked for signals of contamination from other individuals (heterozygosity level of > 60%, Appendix S3, many markers with ambiguous genotypes falling between the genotype clusters and/or conflicting sexing signals), and signs that the sample came from a species other than wolverine (high degree of homozygosity).
SNP genoty** error rates
Precise estimates of genoty** error rates were calculated for 1285 of the monitoring samples (collected 2015–2017) where the true genotype was known from an independent tissue sample or well working monitoring sample (0% marker dropout) of the same individual. “Allele dropout” was defined as markers where the true genotype was heterozygous but the scored genotype was homozygous. “False allele” was defined as all markers where the true genotype was homozygous while the scored genotype was heterozygous. The rare cases where the true genotype was homozygous for one allele while the scored genotype was homozygous for the other allele was scored as both “False allele” and “Allele dropout”. “Marker dropout” was calculated as the rate of non-scored autosomal or X-linked markers. Error rates were calculated independently for each run, thus twice per sample run in duplicate and four times for re-run samples. To avoid pseudo-replication, the mean error rate per sample was used.
In order to evaluate the effect of DNA concentration on genoty** success and genoty** error rates, we performed a dilution series for 8 selected tissue samples. Each sample was diluted four times in water with a ratio of 1:5, resulting in final DNA concentrations of 5 ng/µl, 1 ng/µl, 0.2 ng/µl, 0.04 ng/µl and 0.008 ng/µl. These were then genotyped in duplicate with the same analysis pipeline as other samples. Marker dropout rate, allele dropout rate and false allele rate were calculated as above.
Sexing and individual identification
Genetic sexing of all samples was done using the 6 X-chromosome and 3 Y-chromosome markers included in set D. Genotype cut-offs for sex determination was identified following preliminary genoty** of individuals of known sex. Thus, the sex of a given sample was set to male if the number of positive Y-markers was higher than 1 and the number of heterozygous X-markers was 0. A sample was classified as female if the number of positive Y-markers was 0 and the number of positive X markers was higher than 4, or if the number of positive Y-markers was 0 and the number of heterozygous X markers was higher than 1. In all other cases and for all samples where the marker dropout rate was 25% or higher, the sex was set as “unknown”.
Each sample consensus genotype was matched against a database of all known individual genotypes (as well as all other samples from the same run) using the PLINK (ver. 1.07) –cluster –matrix command (Purcell et al. 2007). The similarity matrix produced was then converted to long format and analysed using a custom R script (supplementary material). All pairwise similarities above 95% (including the same sex assignment) and with more than 85% of markers genotyped in both samples were automatically considered to be multiple samples from the same individual. These cut-offs reliably separate samples from different individuals and samples from the same individual (Ekblom et al. 2018; This study). Samples with similarities between 85% and 95% and those with fewer than 85% of successfully genotyped markers in common were manually checked to confirm identity. For each new identified individual a consensus sequence was produced based on all genotyped samples of that individual and added to the genotype database.
Assessment of number of markers needed for reliable individual identification
In order to evaluate how many SNP markers are needed for reliably separating different individuals based on the multi-locus genotype, we used a total of 182 monitoring samples from 161 different individuals (previously identified from microsatellite analyses, including pairs of individuals known to have high levels of genetic similarity) that were genotyped with all SNPs included in set A and B (thus 173 autosomal and X-linked markers in total). To simulate genoty** with fewer markers, this dataset was reduced by randomly removing different number of markers during the data analysis. The reduced genotypes were then run in the same individual matching pipeline. For each selected number of markers, ten random independent marker sets were thus constructed and analysed. The distributions of the pairwise similarity scores for each sample pair were then compared between samples originating from different individuals and from the same individual. The measure of individual identification success was constructed by taking the difference between the upper 0.5 percentile for the similarity distribution of samples from different individuals and the lower 0.5 percentile for the similarity distribution of samples from the same individual. If this measure is positive, it thus means that less than one percent of the samples have been wrongly inferred to be from the same individual. The probability of mis-assignment was defined as the overlap between the distributions of pairwise similarities for samples from different individuals and from samples from the same individuals, calculated using the R “overlap” function.
Comparison between SNP markers and SSRs
All monitoring samples from 2015 (N = 770) were processed using both the newly developed SNP genoty** pipeline, with marker set D, as well as the analysis procedure previously used for genetic monitoring based on 19 microsatellite (SSR) markers. We could thus compare the performance, in terms of number of successfully genotyped and individually assigned samples, between SSR and SNP markers. SSR markers were amplified in three separate multiplex reactions, fragments were genotyped using an ABI3730xl sequencer and analysed with GeneMapper (Brøseth et al. 2010; Flagstad et al. 2004; Hedmark et al. 2004). In addition, we evaluated the ability to determine the correct sex of samples using the X- and Y-chromosome markers included in SNP set D, compared to the traditional sex identification based on PCR amplification of two DBY intron fragments (DBY3 and DBY7, Hedmark et al. 2004). All samples were run in duplicate, and for cases where these gave inconsistent results the sex of the sample was set to “unknown”.
Pedigree reconstruction
In order to evaluate the performance of SNP genotype data for pedigree reconstruction, we used 84 individuals sampled from the long-term ecological study in Northern Sweden (Sarek samples). For these individuals we had information of known (N = 75; in most cases mother–offspring captured together, but also those identified with SSR kinship analysis with supporting spatial information and age) and assumed (N = 9; based on information about age together with spatial and temporal matching) parent–offspring relationships (Hedmark et al. 2007; Rauset et al. 2015). Most of these were genotyped for all markers used in the A, B and D SNP sets described above (six Sarek individuals were excluded as they lacked identified pedigree relations), as well as the partly overlap** markers from the GoldenGate platform (Illumina) from Ekblom et al. (2018), yielding a total of 357 autosomal markers. No sex chromosome marker genotypes were utilised in the pedigree reconstruction. We thus first constructed the best pedigree possible, using data from all available SNP markers as well as the previously known ecological data. This was then compared to pedigree reconstruction using more limited data sets with fewer SNP-markers (including the 93 markers of SNP set D).
Genetic pedigree reconstruction was carried out using the FRANz software (Riester et al. 2009) using the”full-sib heuristic” algorithm. FRANz input files were built and analysis of the output pedigrees was conducted using R (R Core Team 2016). Pedigrees were visualised using Pedigraph 2.2 (Garbe and Da 2008).
Where a genetically inferred parental relationship matched a known or assumed relationship it was scored as “true”. Where it negated a known or assumed relationship or was impossible due to known birth and death dates it was scored as “false”. All relationships that neither conflicted with, nor were confirmed by, any known relationships were scored as “possible”. To model how the number and accuracy of genetically inferred relationships change with a decreasing number of SNPs we extracted 20 random subsets of different numbers of markers (ranging from 50 to 300) from the 357 SNPs using PLINK 1.07. The full 357 SNP data was also run 20 times to evaluate between-run variation. Concurrence for each parent–offspring relationship was defined as the percentage of the 20 random subsets for each number of markers where the relationship was inferred.
Results
SNP genoty** error rates
We were able to obtain precise estimates of the rates of different kinds of SNP genoty** errors from 1285 of the non-invasively collected monitoring samples. The distribution of genoty** error rates was highly skewed, with a majority of the samples (N = 754) having complete multi-marker profiles with no genoty** errors (Fig. 1). The mean marker dropout rate across all genotypes was 2.7%, while the mean rate of allele dropouts was 1.9% and the mean rate of false alleles was 0.02%. The rates of marker dropout were positively correlated to the rate of genoty** errors, both for allele dropouts (rS = 0.75, df = 1283, p < 0.0001, Fig. 1a), and for false alleles (rS = 0.18, df = 1283, p < 0.0001, Fig. 1b).

Correlation between marker dropout rate and the rate of (a) allele dropouts and (b) false alleles. The distributions of genoty** errors (in the right and top margins of the plots) are highly skewed with a majority of samples overlap** at no marker dropouts and no genoty** errors
The effect of DNA concentration on genoty** success and error rates could be clearly observed by genoty** of a series of diluted tissue samples. Diluted samples with a concentration down to 0.2 ng/µl had complete or near complete genotype profiles, while samples with the lowest concentration of DNA (0.008 ng/µl) showed a marked decrease in genoty** success rate and an elevated level of genoty** errors, especially allele dropouts (Table 1).
Number of SNP markers needed for reliable individual identification
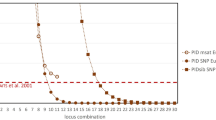
For 182 monitoring samples (161 individuals) we used a total of 173 SNP markers (polymorphic autosomal and X-linked markers from set A, B and D), to evaluate how many SNP markers were needed for making reliable individual assignments. Analyses using 93 markers (as in set D) provided similar power to differentiate genotypes between individuals (probability of mis-assignment <0.001) as using the whole 173 marker set (probability of mis-assignment <0.001, Fig. 2a). Even 45 markers were enough for making reliable individual assignments, based on a very low degree of overlap in pairwise genetic similarities between samples from the same individual and samples from different individuals (probability of mis-assignment = 0.0015). With fewer than 30 markers the ability to differentiate between individuals was reduced (probability of mis-assignment = 0.020), meaning that there is a significant risk that samples may erroneously be inferred to be from different individuals due to genoty** errors, or that samples may erroneously be inferred to come from the same individual due to high genetic similarity between individuals (Fig. 2b). It should be noted that 14 of the samples analysed here came from different individuals that were known to have high levels of relatedness (for example full-sibs from inbred matings; Hedmark and Ellegren 2007), thus representing unusually difficult cases of genetic individual assignment.

Distributions of genetic similarity for pairs of samples depending on the number of markers used (a). Light grey bars indicate genetic similarity of pairs of samples from different individuals while black bars indicate similarity between pairs of samples from the same individual. Dashed vertical lines in each diagram represents the upper 0.5 percentile for the distribution of pairs from different individuals, and solid vertical lines represents the lower 0.5 percentile for pairs from the same individual. Where the dashed line is left of the solid line, the degree of overlap between the two distributions is thus less than 1%. (b) Degree of 1% overlap plotted against the number of markers. A positive overlap indicates that there is less than 1% overlap between the distributions of genetic similarities between pairs of samples from different and the same individuals. Mean and range for 10 independent random subset of markers is shown for runs with fewer than 173 markers
Comparison between SNP markers and SSRs
A total of 770 monitoring samples were processed using both the SNP genoty** pipeline (set D) and the previously used multiplex microsatellite (SSR) genoty** method. The SNP method outperformed the traditional microsatellite method in several ways. Genotypes and individual assignments were obtained for 555 samples (72%) using SNPs, while 476 of the samples (62%) were genotyped and individually assigned using microsatellites (Fig. 3). In two cases, the individual assigned to the sample differed between the two genoty** methods. After manual inspection of SNP and SSR profiles, both of these were concluded to come from manual errors in the microsatellite genotype assignment and were corrected in the database based on the SNP genotype. Another improvement with the SNP genoty** method was that fewer samples (120 [16%] compared to 257 [33%] for microsatellites) needed to be re-run in order to obtain reliable genotypes (Fig. 3).

Genoty** success rate (number of samples genotyped and individually assigned) of the described SNP genoty** pipeline in comparison to the previously utilised microsatellite (SSR) method
The sex of the individual genotyped was assigned using 6 X-linked SNPs and 3 Y-linked monomorphic markers included in set D. Sex was inferred for 523 of the samples (279 females and 244 males). In comparison, 486 of the samples (272 females and 214 males) were sexed using the traditional sex markers based on DBY intron amplification. In one case the inferred sex differed between the two methods, a sample that was identified as female with the traditional markers and male with the new SNP markers. The true sex in this case, was known as male based on other genotyped samples from the same individual.
Pedigree reconstruction
In order to evaluate the ability to reconstruct pedigrees in natural populations using the 93 marker SNP set described here, we first produced the most complete pedigree possible, using all available data (both genetic and ecological). This was then compared to pedigrees produced using reduced sets of SNP markers (including the 93 marker set D). For the 84 individual wolverines in the Sarek samples, we used both the full 357 SNP genotypes and the prior information about age and relatedness, and were able to infer 61 mother–offspring and 40 father–offspring relationships. The full pedigree consisted of up to four consecutive generations (Fig. 4). Most of the genetically reconstructed relationships using SNP-data (50 maternal and 19 paternal) were verified by either known (n = 63) or assumed (n = 6) relationships in the Sarek data. Twelve relationships (7 maternal and 5 paternal) that were previously known, or assumed, could not be verified using the SNP data. All but three of these included at least one individual that was not successfully genotyped in this study. Finally, 20 previously unknown relationships (4 maternal and 16 paternal) were identified using the SNP data (Fig. 4). We only observed one case of close inbreeding in the pedigree, this was a mating between half-siblings and their offspring thus had an inbreeding coefficient of 0.125.

Complete pedigree of 84 wolverines monitored in the Sarek study area, based on FRANz analysis of 357 SNPs with age data, together with previously collected ecological data. Females are represented by ellipses, males by rectangles, grey symbols represent individuals that were not successfully genotyped in this analysis. Green lines represent previously known or inferred relationships that were verified using SNP data. Yellow lines represent previously known or assumed relationships that could not be verified using SNP data. Blue lines represent previously unknown relationships that were inferred using SNP data. The star (*) highlights the only case of close inbreeding found in the pedigree
We used random subsets of SNP markers to evaluate the effect of number of markers used on pedigree reconstruction ability. Here “concurrence” was defined as the percentage of 20 independently inferred pedigrees, constructed with a given number of SNP markers, that contained the relationship in question. The genetically inferred pedigree relationships were verified by comparing with ecological data (see Methods for details). The total number of inferred pedigree relationships increased with reduced number of markers (Fig. 5a). This was an effect of a large number of falsely inferred relationships at a low concurrence (thus only found once or twice out of 20 independent runs) when decreasing the number of SNPs. With many markers, or by applying a high concurrence rate cut-off, the number of false positives was low (Fig. 5b). With 200 or more SNPs, a cut-off level of 50% concurrence was sufficient to eliminate all false relationships, whereas 75% concurrence excluded all negative relationships also with as few as 50 SNPs. With a concurrence rate of 95% the number of correctly inferred “true” relationships dropped when using few markers (<150 SNPs). But with a cut-off concurrence of 75%, even fewer than 100 markers provided reliable results (Fig. 5c).

Number of parent – offspring relationships identified per number of SNP by type of relationship and concurrence level. Colours correspond to different levels of concurrence and curves are fitted with a loess function
Discussion
We describe the development and benchmarking of a SNP genoty** pipeline implemented in the conservation-monitoring program for wolverines in Scandinavia. Our study provides an extensive empirical validation of the use of SNP markers for conservation genetics with non-invasively collected samples that is applicable to many other systems facing similar management challenges. Our 96-marker SNP set consistently outperforms the previous microsatellite/DBY marker panel, providing sufficient information for successfully identifying individuals and sex, and for reconstructing reliable pedigrees.
We found that using 93 SNP markers provided similar power to differentiate genotypes between individuals as using the maximum 173 marker set. While these results may serve as a general starting point also for marker development in other species/populations, we recommend a similar validation process as described here, before adopting a new marker system. Due to the risk of ascertainment bias, the marker panel should be developed and/or validated for the population of interest, since it cannot always be easily transferable to other parts of the distribution range, or related taxa (Clark et al. 2005; Garvin et al. 2010; Helyar et al. 2011; Morin et al. 2004). The number of markers needed will also depend on characteristics of both the population (e.g. relatedness structure and overall levels of genetic diversity) and the markers in question (e.g. minor allele frequency and genoty** error rates).
The SNP set presented here consistently outperforms the 19 microsatellite (SSR) marker panel previously used for wolverine population monitoring, both in terms of a larger proportion of the non-invasive samples genotyped, and in terms of increased precision in individual identification, as a result of reduced genoty** error rates. Fewer samples needed to be reanalysed with SNPs compared to SSRs, and the SNP pipeline thus provides a more time- and cost efficient lab-flow. In addition, sexing previously had to be performed by a separate analysis of Y-chromosome specific PCR fragments (Hedmark et al. 2004). Sex-assignment using the SNP panel outperformed the traditional Y-intron sex-markers in terms of both accuracy and number of samples sexed. Consequently, by including X and Y chromosome markers in the SNP set provided here, the laboratory workflow is simplified by performing the sexing of samples together with the genoty**. It should be noted here that the cost of develo** a novel SNP-panel can be considerable (especially in the absence of a reference genome). The reduced time and cost for genoty** with SNPs must thus be weighed against marker development efforts. Also, as noted above, bi-allelic markers (such as SNPs) may be less transferrable between populations compared to multi-allelic markers such as SSRs.
Importantly, apart from being non-invasively collected samples of low quality, some of the samples analysed here came from individuals that were known to have high levels of relatedness (e.g. full-sibs from inbred matings), and thus representing unusually difficult cases of genetic individual assignment. Consequently, our results highlight the potential applications of similar SNP panels for other study systems. Given that we successfully validated the utility of a SNP marker-set in a population with very low levels of genetic diversity (Ekblom et al. 2018, 2014), SNP markers are expected to be applicable to a wide range of populations with conservation concern, including cases with high levels of inbreeding. An increase in genoty** success rate may be explained by the shorter fragment lengths needed for SNP markers (<120 bp) compared to SSRs (often 150–300 bp), which leads to increased PCR success when using small quantities of degraded DNA (as is often the case in non-invasively collected samples such as scats). The allele-specific signals are also typically clearer for SNP genotypes compared with SSRs, where PCR artefacts such as “stutter-bands” and “primer-dimer peaks” often blur the genotype profiles (Guichoux et al. 2011).
To successfully genotype low quality samples, the SNP genoty** method need to be based on PCR amplification. This limits the potential number of techniques available. We have used the Fluidigm technology, offering genoty** of 96 markers and 96 samples per run. The precise details of our described laboratory procedures and analytical pipeline has been subject to extensive optimisation. For example, we found that using 40 PCR cycles in the pre-amplification (STA) reaction (rather than the 14 suggested by the manufacturer), and diluting the STA product 1:10 (instead of the recommended 1:100), significantly increased genoty** success of low quality samples (see also von Thaden et al. 2020). Further, we achieved the best genotype resolution using 26 cycles (instead of the recommended 34) in the genoty** reaction on the FC1 cycler.
Using DNA samples from free-ranging wolverines from a long-term, individual level ecological study in Northern Sweden, we had the unique opportunity to validate pedigree reconstruction based on SNP genoty** data with prior knowledge of known and assumed relatedness, as well as detailed information on age and the spatial structure (Aronsson and Persson 2018; Rauset et al. 2015). We found that DNA-based pedigree reconstruction was reliable and effective. For a higher number of SNP markers (i.e. ≥150) both the total number of relationships and the number of true relationships identified appear to stabilize. Consequently, it is reasonable to assume that adding more markers (>357) would not increase the accuracy of the reconstructed pedigree. When age data is not available to complement the pedigree, as in most cases using non-invasive sampling, our results suggest that applying a high threshold of concurrence (≥75%) from multiple independent analyses will minimize the inclusion of false relationships (especially wrong direction of parent–offspring relationships) when reconstructing an unknown pedigree.
The use of genetic methodology and non-invasive samples provide a great source of information with huge potential and reduced costs in terms of animal welfare concerns, financial expenditure and sampling effort. Pedigree reconstruction is valuable for long-lived and elusive species in general, where the relative contribution of each individual can be vital to small or isolated populations, and for carnivores in particular, where compromises between conflict mitigation and conservation may lead to low population targets that need to be monitored precisely. In the pedigree reconstruction presented here, we found no evidence of perpetuated matings between closely related individuals among the wolverines in Sarek (only one case of half-sib parentage). There are a few previously described examples of close inbreeding of wolverines in newly formed, small and partly isolated sub-populations in Scandinavia (Hedmark and Ellegren 2007). The Sarek area, however, was highly saturated with wolverines during the entire study period, characterized by a stable distribution of resident individuals (Aronsson and Persson 2018).
The Scandinavian wolverine served as an excellent, challenging, non-model system for benchmarking SNP genoty** in management monitoring, but the methods implemented in this study will be applicable to many other populations and species facing similar challenges. SNP data can also be used to investigate population demography, migration patterns, population structure and effective gene flow (Kleven et al. 2019). Information from such analyses can, in turn, be used for making informed management decisions (regarding for example translocations, population protection, hunting quotas, and protective legislation) thus providing a case for bridging the “conservation genomics gap” (Shafer et al. 2015, 2016). However, the value of genetic data also relies on the accuracy of the prior information required by the programs. Gathering ecological data is still an important and potentially vital part of for example pedigree reconstruction and this cannot be replaced entirely by genetic data. The continued value of ecological data is thus not be underestimated.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Code availability
All software used in this study is outlined throughout the Material and Methods section. Custom scripts are provided in the supplementary material.
References
Åkesson M, Liberg O, Sand H, Wabakken P, Bensch S, Flagstad Ø (2016) Genetic rescue in a severely inbred wolf population. Mol Ecol 25:4745–4756. https://doi.org/10.1111/mec.13797
Anderson EC, Garza JC (2006) The power of single-nucleotide polymorphisms for large-scale parentage inference. Genetics 172:2567–2582. https://doi.org/10.1534/genetics.105.048074
Aronsson M, Persson J (2017) Mismatch between goals and the scale of actions constrains adaptive carnivore management: the case of the wolverine in Sweden. Anim Conserv 20:261–269. https://doi.org/10.1111/acv.12310
Aronsson M, Persson J (2018) Female breeding dispersal in wolverines, a solitary carnivore with high territorial fidelity. Eur J Wildl Res 64:7. https://doi.org/10.1007/s10344-018-1164-3
Aylward ML, Sullivan AP, Perry GH, Johnson SE, Louis EE (2018) An environmental DNA sampling method for aye-ayes from their feeding traces. Ecol Evolut. https://doi.org/10.1002/ece3.4341
Beja-Pereira A, Oliveira R, Alves PC, Schwartz MK, Luikart G (2009) Advancing ecological understandings through technological transformations in noninvasive genetics. Mol Ecol Resour 9:1279–1301. https://doi.org/10.1111/j.1755-0998.2009.02699.x
Bérénos C, Ellis PA, Pilkington JG, Pemberton JM (2014) Estimating quantitative genetic parameters in wild populations: a comparison of pedigree and genomic approaches. Mol Ecol 23:3434–3451. https://doi.org/10.1111/mec.12827
Bischof R, Gregersen ER, Brøseth H, Ellegren H, Flagstad Ø (2016) Noninvasive genetic sampling reveals intrasex territoriality in wolverines. Ecol Evolut 6:1527–1536. https://doi.org/10.1002/ece3.1983
Bischof R, Milleret C, Dupont P, Chipperfield J, Tourani M, Ordiz A, de Valpine P, Turek D, Royle JA, Gimenez O, Flagstad Ø, Åkesson M, Svensson L, Brøseth H, Kindberg J (2020) Estimating and forecasting spatial population dynamics of apex predators using transnational genetic monitoring. Proc Nat Acad Sci 117(48):30531–30538
Brøseth H, Flagstad Ø, Wärdig C, Johansson M, Ellegren H (2010) Large-scale noninvasive genetic monitoring of wolverines using scats reveals density dependent adult survival. Biol Cons 143:113–120
Bruford MW, Davies N, Dulloo ME, Faith DP, Walters M (2017) Monitoring changes in genetic diversity. In: Walters M, Scholes RJ (eds) The GEO handbook on biodiversity observation networks. Springer International Publishing, Cham, pp 107–128
Carroll EL, Bruford MW, DeWoody JA, Leroy G, Strand A, Waits L, Wang J (2018) Genetic and genomic monitoring with minimally invasive sampling methods. Evol Appl 11:1094–1119. https://doi.org/10.1111/eva.12600
Chapron G et al (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519. https://doi.org/10.1126/science.1257553
Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R (2005) Ascertainment bias in studies of human genome-wide polymorphism. Genome Res 15:1496–1502. https://doi.org/10.1101/gr.4107905
Ekblom R et al (2018) Genome sequencing and conservation genomics in the Scandinavian wolverine population. Conserv Biol 32:1301–1312. https://doi.org/10.1111/cobi.13157
Ekblom R, Smeds L, Ellegren H (2014) Patterns of sequencing coverage bias revealed by ultra-deep sequencing of vertebrate mitochondria. BMC Genomics 15:467
Ferreira CM et al (2018) Genetic non-invasive sampling (gNIS) as a cost-effective tool for monitoring elusive small mammals. Eur J Wildlife Res 64:46. https://doi.org/10.1007/s10344-018-1188-8
Flagstad Ø et al (2004) Colonization history and noninvasive monitoring of a reestablished wolverine population. Conserv Biol 18:676–688. https://doi.org/10.1111/j.1523-1739.2004.00328.x-i1
Flagstad Ø et al. (2019) DNA-based monitoring of the Scandinavian wolverine population 2019 vol 1762. Norwegian Institute for Nature Research
Förster DW et al (2018) Targeted resequencing of coding DNA sequences for SNP discovery in nonmodel species. Mol Ecol Resour 18:1356–1373. https://doi.org/10.1111/1755-0998.12924
Garbe JR, Da Y (2008) Pedigraph, A Software Tool for the Graphing and Analysis of Large Complex Pedigree, User Manual Version 2.4. University of Minnesota
Garvin MR, Saitoh K, Gharrett AJ (2010) Application of single nucleotide polymorphisms to non-model species: a technical review. Mol Ecol Resour 10:915–934. https://doi.org/10.1111/j.1755-0998.2010.02891.x
Guichoux E et al (2011) Current trends in microsatellite genoty**. Mol Ecol Resour 11:591–611. https://doi.org/10.1111/j.1755-0998.2011.03014.x
Hedmark E, Ellegren H (2007) DNA-based monitoring of two newly founded Scandinavian wolverine populations. Conserv Genet 8:843–852. https://doi.org/10.1007/s10592-006-9231-9
Hedmark E, Flagstad Ø, Segerström P, Persson J, Landa A, Ellegren H (2004) DNA-based individual and sex identification from wolverine (Gulo Gulo) faeces and urine. Conserv Genet 5:405–410. https://doi.org/10.1023/B:COGE.0000031224.88778.f5
Hedmark E, Persson J, Segerström P, Landa A, Ellegren H (2007) Paternity and mating system in wolverines Gulo gulo. Wildl Biol 13(13–30):18
Helyar SJ et al (2011) Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol Ecol Resour 11:123–136. https://doi.org/10.1111/j.1755-0998.2010.02943.x
Henriksen S, Hilmo O (2015) Norwegian red list of species 2015 – methods and results. Norwegian Biodiversity Information Centre, Norway
Hobbs NT, Andrén H, Persson J, Aronsson M, Chapron G (2012) Native predators reduce harvest of reindeer by Sámi pastoralists. Ecol Appl 22:1640–1654. https://doi.org/10.1890/11-1309.1
Inman RM, Magoun AJ, Persson J, Mattisson J (2012) The wolverine’s niche: linking reproductive chronology, caching, competition, and climate. J Mammal 93:634–644. https://doi.org/10.1644/11-mamm-a-319.1
Johnson WE et al (2010) Genetic restoration of the Florida Panther. Science 329:1641–1645. https://doi.org/10.1126/science.1192891
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Res 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Khan A et al (2020) Are shed hair genomes the most effective noninvasive resource for estimating relationships in the wild? Ecol Evol 10:4583–4594. https://doi.org/10.1002/ece3.6157
Kleven O et al. (2019) Estimation of gene flow into the Scandinavian wolverine vol 1617. Norwegian Institute for Nature Research
Ko A, Nielsen R (2017) Composite Likelihood Method for Inferring Local Pedigrees. bioRxiv. https://doi.org/10.1101/106492
Kraus RHS et al (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour 15:295–305. https://doi.org/10.1111/1755-0998.12307
Marshall TC, Slate J, Kruuk LEB, Pemberton JM (1998) Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7:639–655. https://doi.org/10.1046/j.1365-294x.1998.00374.x
Mattisson J, Rauset GR, Odden J, Andrén H, Linnell JDC, Persson J (2016) Predation or scavenging? Prey body condition influences decision-making in a facultative predator, the wolverine. Ecosphere 7:e01407. https://doi.org/10.1002/ecs2.1407
McMahon SM et al (2011) Improving assessment and modelling of climate change impacts on global terrestrial biodiversity. Trends Ecol Evol 26:249–259. https://doi.org/10.1016/j.tree.2011.02.012
Miller JM, Poissant J, Kijas JW, Coltman DW, the International Sheep Genomics C (2011) A genome-wide set of SNPs detects population substructure and long range linkage disequilibrium in wild sheep. Mol Ecol Res 11:314–322. https://doi.org/10.1111/j.1755-0998.2010.02918.x
Mills LS (2013) Conservation of wildlife populations: demography, genetics and management. 2nd edn. Wiley-Blackwell
Morin PA, Luikart G, Wayne RK, the SNP workshop group (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evolut 19:208–216
Morin PA, Martien KK, Taylor BL (2009) Assessing statistical power of SNPs for population structure and conservation studies. Mol Ecol Resour 9:66–73. https://doi.org/10.1111/j.1755-0998.2008.02392.x
Nilsson T (2013) Population viability analyses of the Scandinavian populations of bear (Ursus arctos), lynx (Lynx lynx) and wolverine (Gulo gulo). Swedish Environmental Protection Agency [Naturvårdsverket]
Norman AJ, Spong G (2015) Single nucleotide polymorphism-based dispersal estimates using noninvasive sampling. Ecol Evolut 5:3056–3065. https://doi.org/10.1002/ece3.1588
Nussberger B, Wandeler P, Camenisch G (2014) A SNP chip to detect introgression in wildcats allows accurate genoty** of single hairs. Eur J Wildl Res 60:405–410. https://doi.org/10.1007/s10344-014-0806-3
Pereira HM et al (2013) Essential biodiversity variables. Science 339:277–278. https://doi.org/10.1126/science.1229931
Persson J (2005) Female wolverine (Gulo gulo) reproduction: reproductive costs and winter food availability. Canadian J Zool 83:1453–1459. https://doi.org/10.1139/z05-143
Persson J, Ericsson G, Segerström P (2009) Human caused mortality in the endangered Scandinavian wolverine population. Biol Cons 142:325–331
Persson J, Rauset GR, Chapron G (2015) Paying for an endangered predator leads to population recovery. Conserv Lett 8:345–350. https://doi.org/10.1111/conl.12171
Persson J, Wedholm P, Segerström P (2010) Space use and territoriality of wolverines (Gulo gulo) in northern Scandinavia. Eur J Wildl Res 56:49–57. https://doi.org/10.1007/s10344-009-0290-3
Purcell S et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Human Genet 81:559–575. https://doi.org/10.1086/519795
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Rauset GR, Low M, Persson J (2015) Reproductive patterns result from age-related sensitivity to resources and reproductive costs in a mammalian carnivore. Ecology 96:3153–3164. https://doi.org/10.1890/15-0262.1
Riester M, Stadler PF, Klemm K (2009) FRANz: reconstruction of wild multi-generation pedigrees. Bioinformatics 25:2134–2139. https://doi.org/10.1093/bioinformatics/btp064
Rodgers TW, Janečka JE (2013) Applications and techniques for non-invasive faecal genetics research in felid conservation. Eur J Wildl Res 59:1–16. https://doi.org/10.1007/s10344-012-0675-6
SEPA (2014) National management plan for wolverines – management period 2014–2019. Swedish Environmental Protection Agency
Shafer ABA et al (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87. https://doi.org/10.1016/j.tree.2014.11.009
Shafer ABA et al (2016) Reply to Garner. Trends Ecol Evolut 31:83–84. https://doi.org/10.1016/j.tree.2015.11.010
Snyder-Mackler N et al (2016) Efficient genome-wide sequencing and low-coverage pedigree analysis from noninvasively collected samples. Genetics 203:699–714. https://doi.org/10.1534/genetics.116.187492
Soulé ME (1985) What is conservation biology? Bioscience 35:727–734. https://doi.org/10.2307/1310054
Soulé ME, Mills LS (1992) Conservation genetics and conservation biology: a troubled marriage. Conservation of biodiversity for sustainable development. Scandinavian University Press, Oslo, pp 55–69
Spitzer R, Norman AJ, Schneider M, Spong G (2016) Estimating population size using single-nucleotide polymorphism-based pedigree data. Ecol Evolut 6:3174–3184. https://doi.org/10.1002/ece3.2076
Staples J, Qiao D, Cho Michael H, Silverman Edwin K, Nickerson Deborah A, Below Jennifer E (2014) PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am J Human Genet 95:553–564. https://doi.org/10.1016/j.ajhg.2014.10.005
Swedish species information centre (2015) Red listed species in Sweden. Swedish Agricultural University, Uppsala
USFWS (2013) U.S. Federal Register, Washington, D.C., USA. https://www.federalregister.gov/articles/2013/02/04/2013-01478/.
Vilà C et al (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc Royal Soc London Ser B Biol Sci 270:91–97. https://doi.org/10.1098/rspb.2002.2184
von Thaden A et al (2020) Applying genomic data in wildlife monitoring: development guidelines for genoty** degraded samples with reduced single nucleotide polymorphism (SNP) panels. Mol Ecol Res. https://doi.org/10.1111/1755-0998.13136
Walker CW, Vilà C, Landa A, Lindén M, Ellegren H (2001) Genetic variation and population structure in Scandinavian wolverine (Gulo gulo) populations. Mol Ecol 10:53–63. https://doi.org/10.1046/j.1365-294X.2001.01184.x
Wang J, Santure AW (2009) Parentage and sibship inference from multilocus genotype data under polygamy. Genetics 181:1579–1594. https://doi.org/10.1534/genetics.108.100214
Acknowledgements
We thank field workers and local administrative boards for performing many hours of field work and providing samples, especially Peter Segerström for his leading role in sampling data from wolverines captured within the Sarek area (The Swedish Wolverine Project). H Ellegren for providing data and feedback on earlier versions of this manuscript. Ø Flagstad for sharing samples from Norway and SVA (National Veterinary Institute) for tissue samples. J Magnusson assisted in lab work. High performance computing was conducted using the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Funding
Open access funding provided by The Swedish Environmental Protection Agency. The Swedish Environmental Protection Agency to RE. The Swedish wolverine project in Sarek was funded by the Swedish Environmental Protection Agency to JP, Norwegian Directorate for Nature Management, Swedish Research Council Formas, World Wildlife Fund Sweden, European Association of Zoos and Aquaria, and Marie-Claire Cronstedt Foundation.
Author information
Authors and Affiliations
Contributions
RE wrote the manuscript and performed analyses together with MA, FEG, MJ, TF and JP. RE and JP conceived the study. RE and JP obtained funds for the project. MJ and RE performed laboratory work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Captures, handling, and tissue sampling from live animals were approved by the Swedish Ethical Committee on Animal Research and the Swedish Environmental Protection Agency.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ekblom, R., Aronsson, M., Elsner-Gearing, F. et al. Sample identification and pedigree reconstruction in Wolverine (Gulo gulo) using SNP genoty** of non-invasive samples. Conservation Genet Resour 13, 261–274 (2021). https://doi.org/10.1007/s12686-021-01208-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-021-01208-5