
Abstract
Background
Trichohepatoenteric syndrome (THES) is characterized by neonatal-onset intractable diarrhea. It often requires long-term total parenteral nutrition (TPN). In addition, other characteristic findings of the syndrome include growth retardation, facial dysmorphism, hair abnormalities, various immunological problems and other rare system findings. Two genes and their associated pathogenic variants have been associated with this syndrome: SKIC3 and SKIC2.
Methods and results
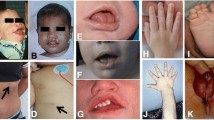
In this case series, the clinical findings and molecular analysis results of a total of 8 patients from 5 different families who presented with persistent diarrhea and were diagnosed with THES were shared. Pathogenic variants were detected in the SKIC3 gene in 6 of our patients and in the SKIC2 gene in 2 patients. It was planned to compare the clinical findings of our patients with other patients, together with literature data, and to present yet-undefined phenotypic features that may be related to THES. In our case series, in addition to our patients with a novel variant, patient number 2 had a dual phenotype (THES and Spondyloepimetaphyseal dysplasia, sponastrime type) that has not been reported yet. Delay in gross motor skills, mild cognitive impairment, radioulnar synostosis, osteoporosis, nephropathy and cystic lesions (renal and liver) were observed as unreported phenotypic findings.
Conclusions
We are expanding the clinical and molecular repertoire of the syndrome regarding patients diagnosed with THES. We recommend that the NGS (next-generation sequencing) multigene panel should be used as a diagnostic tool in cases with persistent diarrhea.




Similar content being viewed by others

Data availability
The data used to support the findings of this study are available from the corresponding author upon reasonable request.
References
Taher ZA et al (2020) A new variant mutation in SKIV2L Gene in Case of Trichohepatoenteric Syndrome. Pediatr Rep 12(3):93–97
Verloes A et al (1997) Tricho-Hepato-enteric syndrome: further delineation of a distinct syndrome with neonatal hemochromatosis phenotype, intractable diarrhea, and hair anomalies. Am J Med Genet 68(4):391–395
Baynam G et al (2017) Initiating an undiagnosed diseases program in the western Australian public health system. Orphanet J Rare Dis 12(1):83
Hartley JL et al (2010) Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology 138(7):2388–2398
Lee WS et al (2016) Novel mutations in SKIV2L and TTC37 genes in Malaysian children with trichohepatoenteric syndrome. Gene 586(1):1–6
Girault D et al (1994) Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency. J Pediatr 125(1):36–42
Hiejima E et al (2017) Tricho-Hepato-enteric syndrome with novel SKIV2L gene mutations: a case report. Med (Baltim) 96(46):e8601
Fabre A et al (2012) SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet 90(4):689–692
Goulet O et al (2008) Syndromic (phenotypic) diarrhea in early infancy. Orphanet J Rare Dis 3:6
Bourgeois P et al (2018) Tricho-Hepato-Enteric syndrome mutation update: mutations spectrum of TTC37 and SKIV2L, clinical analysis and future prospects. Hum Mutat 39(6):774–789
Fabre A et al (2013) Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis 8(1):5
Thaventhiran JED et al (2020) Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 583(7814):90–95
**nias I et al (2018) Trichohepatoenteric syndrome: a rare mutation in SKIV2L gene in the first Balkan reported case. SAGE Open Med Case Rep 6:2050313x18807795
Barabino AV et al (2004) Syndromic diarrhea may have better outcome than previously reported. J Pediatr 144(4):553–554
Vély F et al (2018) Combined immunodeficiency in patients with Trichohepatoenteric Syndrome. Front Immunol 9:1036
Berni Canani R et al (2010) Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr 50(4):360–366
Schaeffer D et al (2010) Functions of the cytoplasmic exosome RNA exosome, : pp. 79–90
Lee KY et al (2024) Long term outcomes in children with trichohepatoenteric syndrome. Am J Med Genet A 194(2):141–149
Kotecha U et al (2012) Trichohepatoenteric syndrome: founder mutation in Asian indians. Mol Syndromol 3(2):89–93
Fabre A et al (2018) A new mutation in the C-terminal end of TTC37 leading to a mild form of syndromic diarrhea/tricho-hepato-enteric syndrome in seven patients from two families. Am J Med Genet A 176(3):727–732
Stankler L et al (1982) Unexplained diarrhoea and failure to thrive in 2 siblings with unusual facies and abnormal scalp hair shafts: a new syndrome. Arch Dis Child 57(3):212–216
Rider NL et al (2015) Novel TTC37 mutations in a patient with immunodeficiency without Diarrhea: extending the phenotype of Trichohepatoenteric Syndrome. Front Pead, 3(2)
Fabre A et al (2014) Syndromic (phenotypic) diarrhoea of infancy/tricho-hepato-enteric syndrome. Arch Dis Child 99(1):35–38
Hosking LM et al (2018) Trichohepatoenteric Syndrome presenting with severe infection and later Onset Diarrhoea. J Clin Immunol 38(1):1–3
Dorum S, Gorukmez O (2021) Expanding the clinical spectrum in trichohepatoenteric syndrome. Am J Med Genet A 185(10):2873–2877
Poulton C et al (2019) Tricho-hepatic-enteric syndrome (THES) without intractable diarrhoea. Gene 699:110–114
Yang M, Jiang Y, Shao X (2022) Case Report: a Novel homozygous frameshift mutation of the SKIV2L Gene in a Trichohepatoenteric Syndrome Patient presenting with short stature, premature ovarian failure, and osteoporosis. Front Genet 13:879899
Fabre A, Badens C (2014) Human mendelian diseases related to abnormalities of the RNA exosome or its cofactors. Intractable Rare Dis Res 3(1):8–11
Houseley J, Tollervey D (2009) The many pathways of RNA degradation. Cell 136(4):763–776
Lebreton A, Séraphin B (2008) Exosome-mediated quality control: substrate recruitment and molecular activity. Biochim Biophys Acta 1779(9):558–565
Mitchell P et al (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3’-->5’ exoribonucleases. Cell 91(4):457–466
Staals RH, Pruijn GJ (2011) The human exosome and disease. Adv Exp Med Biol 702:132–142
Eckard SC et al (2014) The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 15(9):839–845
Rice GI et al (2014) Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46(5):503–509
Kirou KA et al (2005) Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 52(5):1491–1503
Acknowledgements
We thank the patients and families for their participation in this study. We also appreciate the assistance of medical doctors and nurses during sample collection. Each author named in the article has reviewed the article, approved the submission of this version of the article, and assumes full responsibility for the article.
Funding
No funds, grants, or other support was received.
Author information
Authors and Affiliations
Contributions
Each author named in the article has reviewed the article, approved the submission of this version of the article, and assumes full responsibility for the article. All clinicians (MO, KA, ZE, HM, CA, KB, AG, MD, HS, OFB, FIV, IKG, RO and IT) had important roles in the data collection and interpretation of these patients.
Corresponding author
Ethics declarations
Ethical approval
This study was approved by Inonu University Faculty of Medicine Non-Invasive Clinical Research Ethics Committee (approval no.: 2024/5818). The study was conducted following the guidelines and regulations outlined by the relevant ethics committees and institutional review boards. Informed consent was obtained from the parents of the patients described.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ozturk, M., Ates, K., Esener, Z. et al. Expanding the phenotypic and genotypic characteristics of trichohepatoenteric syndrome: a report of eight patients from five unrelated families. Mol Biol Rep 51, 736 (2024). https://doi.org/10.1007/s11033-024-09656-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09656-6