
Abstract
COVID-19 has shaken all the countries across the globe and researchers are trying to find promising antiviral to cure the patients suffering from infection and can decrease the death. Even, different nations are using repurposing drugs to cure the symptoms and these repurposing drugs are hydroxychloroquine, remdesivir, and lopinavir, and recently, India has recently given the approval for the 2-deoxy-d-glucose for emergency purpose to cure the patients suffering from the COVID-19. Plitidepsin is a popular molecule and can be used in treatment of myeloma. Plitidepsin was explored by scientists experimentally against the COVID-19 and was given to the patient. It is found to be more a promising repurposing drug against the COVID-19 than the remdesivir. Therefore, there is a need to understand the interaction of plitidepsin with the main protease of SARS-CoV-2. Molecular docking of the plitidepsin against Mpro of SARS-CoV-2 was performed and the binding energy was found to be − 137.992 kcal/mol. Furthermore, authors have performed the molecular dynamics simulations of the main protease of SARS-CoV-2 in presence of plitidepsin at 300 and 325 K. It was found that the plitidepsin binds effectively with the main protease of SARS-CoV-2 at 300 K.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first pneumonia cases of unknown origin were identified in Wuhan, the capital city of Hubei province, in early December 2019. Coronavirus disease 2019 (COVID-19) has been declared a public health emergency of international concern by the World Health Organization (WHO) (Bojkova et al. 2020; Kumar et al. 2020a, b; Vargas 2020; Saraswat et al. 2021). The pathogen has been identified as a novel enveloped RNA beta-coronavirus known as severe acute respiratory syndrome virus (SARS) coronavirus type 2 (SARS-CoV-2) which has a phylogenetic relationship with SARS-CoV. Since the SARS outbreak about 18 years ago, a large number of SARS-related coronaviruses (SARS-CoV) have been discovered in bats, their natural reservoir host (Hagar et al. 2020; Wu et al. 2020; Peeri et al. 2021). Previous research already suggested that some of the bat SARS-CoVs could infect humans. At the start of the outbreak, full-length genome sequences were obtained from five patients. They are nearly identical and share 79.5% sequence identity with SARS-CoV. Furthermore, it was discovered that nCoV-2019 is 96% identical to a bat coronavirus at the whole-genome level. The surface spike (S) glycoprotein of the SARS-CoV-2 virion is required for host cell receptor binding and is thought to be a key determinant of host range restriction. The SARS-CoV-2 includes 14 ORFs, which are used to encode several proteins. It is made up of ORF, envelope (E) gene, spike (S) gene, nucleocapsid (N) gene, membrane (M) gene, 3’UTR and 5’UTR, etc. MERS-CoV are nearly linked with SARS-CoV-2 because of similar genome (Vargas 2020; Wu et al. 2020; Peeri et al. 2021). Although, SARS-CoV and MERS-CoV are less infectious than the SARS-CoV-2 because of the unlike epidemiological gestures (Vishvakarma et al. 2020; Peeri et al. 2021; Pandamooz et al. 2022). Plitidepsin is called a cyclic depsipeptide that can be first isolated from the Mediterranean marine tunicate, but now, it can be manufactured synthetically commercialized as aplidin. Due to promising biological anti-cancerous ability, it is under trials. Its molecular formula is C57H87N7O15 with molecular weight of 1110.34. Plitidepsin can be prepared in lyophilized dosage, because it degrades under the heat and light stress testing condition. Plitidepsin is supplied as a powder to form a concentrated solution for the confusion in at concentration 2 mg/vial. The observed solution is colorless and having the visible particles. Plitidepsin not only effect the malignant B-CLL but also effective toward the potency of nurse like cells and monocytes that cause leukemia cell progression. It can be studied by the researchers that eukaryotic elongation factor 1A2(eEF1A2) is the primary target of Plitidepsin that can be over expressed in tumors contains multiple myeloma, prostate, pancreas, and ovarian cancers. Therefore, it can be seen that eEF1A2 is an important target for the cancer treatment and biomarker predicting drug sensitivity (Geoerger et al. 2012; Leisch et al. 2019; Gomes et al. 2020; Martinez 2021; White et al. 2021). During the protein synthesis in the ribosome translational elongation factors of EF1A, it participates in the targeting and effective binding of the cognate aminoacyl-tRNA to the acceptor site. EF1A is also known as EF-1alpha. Conformational changes occur in EF1A and it simulates GTP hydrolysis. It results in the detachment of EF1A-GDP from ribosomes and leaving aminoacyl-tRNA attached at acceptor site (Soto-Matos et al. 2011; Barboza et al. 2012; Geoerger et al. 2012; Ribrag et al. 2013; Losada et al. 2016; Leisch et al. 2019; Gomes et al. 2020). The study of interaction between the plitidepsin and eEF1A2 shows that plitidepsin shows the interaction with GTP bound eEF1A2 at nanomolar ranges at different concentration shows the proapoptotic effect in tumor cells; eEF1A2 only protein that can be obtained from the tumor cells lysate, eEF1A2 is decreased in plitidepsin-resistant cells which again can be synthesized in the compounds. In vitro combination studies showed that the when the plitidepsin is combined with different anitcancerous agents like dexamethasone, lenalidomide bortezomib in the MM cell lines; while in the vivo studies showed that plitidepsin shows the antitumor effects in MM xenograft models, T-cell lymphoma, Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL), and pancreatic cancers. It shows the toxicological profile of plitidepsin that is characteristic of a cytotoxic drug for which the liver, spleen, gastrointestinal tract, and bone marrow are the main target organs. Plitidepsin that is hydrophobic drug is now developed using cremophor that causes the unwanted hypersensitivity reactions (Alonso-Álvarez et al. 2017; Leisch et al. 2019; Martinez 2021; White et al. 2021). Molecular docking techniques aim to predict the best matching binding mode of a ligand to a macromolecular partner (here just proteins are considered). It consists in the generation of a number of possible conformations/orientations, i.e., poses, of the ligand within the protein binding site. For this reason, the availability of the three-dimensional structure of the molecular target is a necessary condition; it can be an experimentally solved structure or a structure obtained by computational techniques. Molecular docking is composed mainly by two stages: an engine for conformations/orientations sampling and a scoring function, which associates a score to each predicted pose (Hagar et al. 2020; Vishvakarma et al. 2020). The sampling process should effectively search the conformational space described by the free energy landscape, where energy, in docking, is approximated by the scoring function. The scoring function should be able to associate the native bound conformation to the global minimum of the energy hypersurface. Molecular docking is a key tool in structural molecular biology and computer-assisted drug design. The goal of ligand–protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure. Successful docking methods search high-dimensional spaces effectively and use a scoring function that correctly ranks candidate dockings. Docking can be used to perform virtual screening on large libraries of compounds, rank the results, and propose structural hypotheses of how the ligands inhibit the target, which is invaluable in lead optimization (Kumar et al. 2019, 2020a; Hagar et al. 2020). The setting up of the input structures for the docking is just as important as the docking itself, and analyzing the results of stochastic search methods can sometimes be unclear. This chapter discusses the background and theory of molecular docking software, and covers the usage of some of the most-cited docking software (Hsu et al. 2011). Molecular dynamics simulations analyze the physical movements of atoms and the molecules. These molecules and atoms are interacted in the fixed periods of times that give the dynamic view of the system. Atoms and molecules trajectories are determined using Newton’s equation of motion of the molecules that can be interacted and different forces between the atoms and molecules and the potential energies. These types of methods are applied in chemical physics, material science, and biophysics (Kumar et al. 2019; Vishvakarma et al. 2019; Meena et al. 2020, 2021). Plitidepsin was explored by scientists experimentally against the COVID-19 and was given to the patient. It is found to be more a promising repurposing drug against the COVID-19 than the remdesivir (Martinez 2021; White et al. 2021).
In the present work, authors have investigated the potential of plitidepsin for the inhibition of main protease of SARS-CoV-2 using molecular dynamics simulations at different temperature. Furthermore, the structure of plitidepsin was studied using density functional approach in gaseous state at temperature.
Theoretical calculations
Designing of the ligands
The structure of plitidepsin with stereochemistry, and its three-dimensional structure were drawn using the chemdraw as in Fig. 1, and then, it was optimized using the MM2 calculation available in chemdraw. The crystal structure of the Mpro of the SARS-CoV-2 was taken from the RCSB with PDB ID of 6LU7. Then, the structure of protein was prepared using Chimera for the molecular docking and the molecular dynamics simulations. Furthermore, the change in the structure of Mpro of the SARS-CoV-2 in the presence of plitidepsin was studied using the molecular dynamics (MD) simulations at 300 K and 325 K.

a Two-dimensional structure and b three-dimensional structure of plitidepsin [IUPAC name of plitidepsin is (2S)-N-[(2R)-1-[[(3S,6S,8S,12S,13R,16S,17R,20S,23S)-13-[(2S)-butan-2-yl]-12-hydroxy-20-[(4-methoxyphenyl) methyl]-6,17,21-trimethyl-3-(2-methylpropyl)-2,5,7,10,15,19,22-heptaoxo-8-propan-2-yl-9,18-dioxa-1,4,14,21-tetrazabicyclo[21.3.0]hexacosan-16-yl]amino]-4-methyl-1-oxopentan-2-yl]-N-methyl-1-(2-oxopropanoyl)pyrrolidine-2-carboxamide]
Molecular docking
The structure of Mpro of SARS-CoV-2 has been taken from RCSB and was prepared using Chimera to avoid errors before performing the molecular docking and molecular dynamics simulation. Molecular docking of the plitidepsin with Mpro of nCoV was performed using iGemDock with high accuracy (Vishvakarma et al. 2015; Kumar et al. 2019, 2020d; Mishra et al. 2021). The selected population size was 800 with 80 generations. It provides the binding energy for the formation of the complex between the plitidepsin and the Mpro of SARS-CoV-2 (Kumar et al. 2019, 2020b; Vishvakarma et al. 2019; Meena et al. 2020).
Molecular dynamics simulations
Molecular dynamics simulations of Mpro of SARS-CoV-2 in the presence of plitidepsin was performed using the online WEBGRO Macromolecular Simulations server (https://simlab.uams.edu/ProteinWithLigand/protein_with_ligand.html). It is made available free to the researchers and academicians throughout the world for a better cause. It is a GRACE High-Performance Computing Facility and made available by the University of Arkansas for Medical Sciences (UAMS) as a public service. Before performing MD simulations, there is a need to create the topology of the plitidepsin, that is, the molecular of interest using the Prodrug, that is, GlycoBioChemPRODRG2 server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg). It is an online server. The force filed applied for the MD simulations GORMACS43a1 for Mpro with plitidepsin along with SPS water model in triclinic system and sodium chloride. Then, the energy of the complex formed was minimized using the steepest descent integrator at every 5000 steps. The equilibration NVT/ NPT was performed at 300 K and 1 bar pressure. Further, the MD integrator taken was Leap-frog for a simulation time of 100 ns, as it was limitation and the frames per MD simulations fixed to 1000 (Lindorff-Larsen et al. 2010)(Lindahl et al. 2010)(Oostenbrink et al. 2004; Schüttelkopf and Van Aalten 2004). MD simulations gave various trajectories like the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bonds (HBs) to understand the formation of complex at 300 K and 325 K (Lindahl et al. 2010; Abraham et al. 2015).
Results and discussion
Molecular docking
Herein, dock prep module was used to check the following parameters, i.e., adding hydrogen, removal of solvents, replacing incomplete residues and charges were assigned. Molecular docking of the plitidepsin was performed against the Mpro of nCoV using iGemDock with high accuracy as mentioned in experimental. The selected population size was 800 with 80 generations and 10 solutions. It provides the binding energy as well as interaction energy of particular amino acids of the active binding cavity for the formation of the complex between the plitidepsin and the Mpro of SARS-CoV-2.
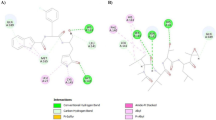
The binding energy for the formation of the complex between the plitidepsin and the Mpro of SARS-CoV-2 was found as − 137.992 kcal/mol. It is due to the contribution made by energy of − 111.365 and − 26.627 kcal/mol of van der Waal’s interaction and hydrogen bonding respectively. It does not show electrostatic interaction. The docked pose against the Mpro of SARS-CoV-2 in 2D and 3D is mentioned in Fig. 2. It can be seen that the plitidepsin fits into the active cavity and interacts effectively with the amino acids of Mpro of SARS-CoV-2 (Table 1).

a 3D- and b 2D-docked view of plitidepsin with the Mpro of SARS-CoV-2
Plitidepsin has been docked against the Mpro of nCoV and 10 poses have been collected to understand the importance of receptor site and the amino acids involved in interaction. Total number of 10 poses were obtained on interaction at four sites. Mostly binds to same site as pose number 4 is obtained. It gave best binding energy in kcal/mol.
Furthermore, Table 2 has provided the information about the interaction of plitidepsin with the amino acids of Mpro of SARS-CoV-2. Plitidepsin forms classical hydrogen bonds with the ASN-238 (2.39 Å), THR-199 (2.22 Å), LYS-137 (2.81 Å), and ASP-197 (2.53 Å). Furthermore, it forms non-classical hydrogen bonds with the LEU-286, THR-198, and ASP-289. It shows hydrophobic interaction with the LEU-286, LEU-287, and LYS-137.
The binding cavity for amino acid residue contribution toward the stabilization of plitidepsin was analyzed by iGEMDOCK. Binding energy versus contributing amino acid residues of active cavity is plotted as given in Fig. 3. The major contributing amino acid residues of active cavity for the Plitidepsin are LYS-137, ASP-197, THR-199, LEU-286, ASP-289, ARG-131, and LEU-287, in which the LYS-137 have highest negative contribution, which corroborate the successful docking.

Binding energy contributions of the amino acids of active cavity of Mpro of nCoV
Molecular dynamics simulations
Molecular dynamic simulation is a technique to simulate the whole protein–ligand system due to course of certain time to analyze the confirmation changes (Lindorff-Larsen et al. 2010). It applies the Newton’s law of motion to the macromolecular system and generates trajectories for the given time period of simulation. By analyzing the coordinates of trajectories points, certain parameters can be analyzed to define the stability of protein–ligand complex (Kumar et al. 2019, 2020c; Vishvakarma et al. 2019; Meena et al. 2021). These parameters are radius of gyration, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and number of hydrogen bonds. Various trajectories like Rg, RMSD, hydrogen bond, and RMSF of the main protease of SARS-CoV-2 in presence of plitidepsin at 300 and 325 K were drawn. Overall changes in the structure of the main protease of SARS-CoV-2 are mainly investigated by the RMSD’s of the main chain atoms of the protease due to a molecule from the initial places or coordinates of the atoms of the prepares protein. It can also be explored by studying the radius of gyration of the protein in small-angle X-ray scattering studies.
Radius of gyration (Rg) is a parameter to measure the strength of the macromolecular system using trajectories points obtained from MD simulations. It is the distance between the axis of rotation and the center of mass. Conformational stability in term of Rg can be described as, if there is less fluctuations in Rg values occur indicates more conformational stability of the protein–ligand system or complex. Less the average value of Rg indicate more compact system. Figure 4 can be used to understand the radium of gyration of the complex of plitidepsin with Mpro of SARS-CoV-2 at 300 and 325 K. From this, it can be seen the deviation is more in case of trajectory obtained at 325 K than 300 K obtained from the MD simulations. In the beginning, that is, at about 15 ns, the deviation at 300 K is found more, but on running MD simulations for 100 ns, the fluctuations are less at 300 K than in the complex at 325 K. It can be said that the inhibition will be more at 300 K.

Radius of gyration for the complex between plitidepsin with main protease of SARS-CoV-2 at 300 and 325 K
Hydrogen bonds is considered as strong dipole–dipole interaction. It has two types, conventional and non-conventional. Conventional hydrogen bonds found among hydrogen and N, O, F (nitrogen, oxygen, fluorine) atoms, while no-conventional hydrogen bonds found among hydrogen and other elements other than N, O, F. Hydrogen bonds and its number are more important for the protein–ligand complex. The stability of ligand into the active binding cavity of protein is measured in terms of average number of hydrogen bonds. The length of interaction is more useful, shorter the length stronger will be interaction. Figure 5 is used to study the number of hydrogen bonds in the complex of plitidepsin with main protease of SARS-CoV-2 at 300 and 325 K. It can be seen that a greater number of hydrogen bonds are observed in the trajectory obtained at 300 K than the 325 K. At 300 K, a maximum number of hydrogen bonds are eight, while at 325, it decreases and the maximum number of hydrogen bonds is seven. As known, the hydrogen bond between the ligand and receptor is an important parameter for the inhibition of the main protease of SARS-CoV-2 using plitidepsin.

Hydrogen bonds for the complex between plitidepsin with main protease of SARS-CoV-2 at 300 and 325 K
Root-mean-square deviation is another way to analyze the compactness of protein after the ligand-induced fit into it (Lindorff-Larsen et al. 2010; Abraham et al. 2015). In this, atomic coordinates of backbone atoms from the trajectories were obtained to measure the RMSD. It is calculated by rooting the average value of square of these atomic coordinates. Less the fluctuation in the RMSD values of the complex indicates greater stability. RMSD value within 3 angstrom indicated successful docking or fit. There is need to discuss the RMSD of the main protease of SARS-CoV-2 with plitidepsin (Fig. 6). From the plots, it can be understood that at high temperature, the RMSD values of the complex are much higher at 325 K on comparing with the RMSD obtained at 300 K. At 325 K, the RMSD values reach to 5 angstrom, while the RMSD values at 300 K are less than 3 angstrom and aceeptable.

RMSD fits to backbone for the complex between plitidepsin with main protease of SARS-CoV-2 at 300 and 325 K
Root-mean-square fluctuation (RMSF) is also useful to define the conformational stability of the macromolecular system. It is calculated in the same approach as for the RMSD, but herein, it uses individual residue flexibility. Less the fluctuation of coordinates indicates the more stability. A symmetrical fluctuation indicates greater stability compared to unsymmetrical fluctuation. When RMSF values of protein and protein–ligand complex were plotted along with the fluctuation in some part, it indicates the induced fitting of ligand. From Fig. 7, it is observed that the fluctuation in more in case of MD simulations is performed at 325 K in comparison of 300 K. Therefore, the complex is more stable at 300 K, and so, the inhibition of main protease of SARS-CoV-2 is more at 300 K.

RMSF for the complex between plitidepsin with main protease of SARS-CoV-2 at 300 and 325 K
Conclusion
Use of plitidepsin against the COVID-19 is the development of antiviral drugs may be acceptable. Plitidepsin is reported to be a more promising candidate against the SARS-CoV-2 infection than remdesivir. Herein, the potential of plitidepsin was investigated for the inhibition of the Mpro of SARS-CoV-2. Molecular docking of the plitidepsin against Mpro of SARS-CoV-2 was performed and the binding energy in best pose out of 10 poses was found to be − 137.992 kcal/mol. To find the binding through the complex of plitidepsin with main protease of SARS-CoV-2 was studied through the molecular dynamics simulations at 300 K. Furthermore, the binding was studied at higher temperature, that is, at 325 K. It was found that the plitidepsin binds effectively with the main protease of SARS-CoV-2 at 300 K.
References
Abraham MJ, Murtola T, Schulz R et al (2015) Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. https://doi.org/10.1016/j.softx.2015.06.001
Alonso-Álvarez S, Pardal E, Sánchez-Nieto D et al (2017) Plitidepsin: design, development, and potential place in therapy. Drug Des Devel Ther 11:253
Barboza NM, Medina DJ, Budak-Alpdogan T et al (2012) Plitidepsin (Aplidin) is a potent inhibitor of diffuse large cell and Burkitt lymphoma and is synergistic with rituximab. Cancer Biol Ther. https://doi.org/10.4161/cbt.13.2.18876
Bojkova D, Costa R, Bechtel M et al (2020) Targeting pentose phosphate pathway for SARS-CoV-2 therapy. BioRxiv 11:699
Geoerger B, Estlin EJ, Aerts I et al (2012) A phase i and pharmacokinetic study of plitidepsin in children with advanced solid tumours: an innovative therapies for children with cancer (ITCC) study. Eur J Cancer. https://doi.org/10.1016/j.ejca.2011.10.036
Gomes NGM, Valentaõ P, Andrade PB, Pereira RB (2020) Plitidepsin to treat multiple myeloma. Drugs Today. https://doi.org/10.1358/dot.2020.56.5.3135886
Hagar M, Ahmed HA, Aljohani G, Alhaddad OA (2020) Investigation of some antiviral N-heterocycles as COVID 19 drug: molecular docking and DFT calculations. Int J Mol Sci. https://doi.org/10.3390/ijms21113922
Hsu KC, Chen YF, Lin SR, Yang JM (2011) Igemdock: a graphical environment of enhancing gemdock using pharmacological interactions and post-screening analysis. BMC Bioinform. https://doi.org/10.1186/1471-2105-12-S1-S33
Kumar D, Singh P, Jayaraj A et al (2019) A theoretical model to study the interaction of erythro-noscapines with nsp3 protease of chikungunya virus. ChemistrySelect. https://doi.org/10.1002/slct.201803360
Kumar A, Kumar D, Kumar R et al (2020a) DFT and docking studies of designed conjugates of noscapines & repurposing drugs: promising inhibitors of main protease of SARS-CoV-2 and falcipan-2. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1841030
Kumar D, Kumari K, Jayaraj A et al (2020b) Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1752310
Kumar D, Singh P, Jayaraj A et al (2020c) Selective docking of pyranooxazoles against nsP2 of CHIKV eluted through isothermally and non-isothermally MD simulations. ChemistrySelect. https://doi.org/10.1002/slct.202000768
Kumar RV, Srivastava D, Singh V et al (2020d) Characterization, biological evaluation and molecular docking of mulberry fruit pectin. Sci Rep. https://doi.org/10.1038/s41598-020-78086-8
Leisch M, Egle A, Greil R (2019) Plitidepsin: a potential new treatment for relapsed/refractory multiple myeloma. Futur Oncol 15:109
Lindahl E, Bjelkmar P, Larsson P et al (2010) Implementation of the charmm force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theory Comput. https://doi.org/10.1021/ct900549r
Lindorff-Larsen K, Piana S, Palmo K et al (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct Funct Bioinform. https://doi.org/10.1002/prot.22711
Losada A, Muñoz-Alonso MJ, García C et al (2016) Translation elongation factor eEF1A2 is a novel anticancer target for the marine natural product plitidepsin. Sci Rep. https://doi.org/10.1038/srep35100
Martinez MA (2021) Plitidepsin: a repurposed drug for the treatment of COVID-19. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00200-21
Meena MK, Kumar D, Jayaraj A et al (2020) Designed thiazolidines: an arsenal for the inhibition of nsP3 of CHIKV using molecular docking and MD simulations. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1832918
Meena MK, Kumar D, Kumari K et al (2021) Promising inhibitors of nsp2 of CHIKV using molecular docking and temperature-dependent molecular dynamics simulations. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1873863
Mishra D, Maurya RR, Kumar K et al (2021) Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J Mol Liq. https://doi.org/10.1016/j.molliq.2021.116185
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. https://doi.org/10.1002/jcc.20090
Pandamooz S, Jurek B, Meinung C-P et al (2022) Experimental models of SARS-CoV-2 infection: possible platforms to study COVID-19 pathogenesis and potential treatments. Annu Rev Pharmacol Toxicol. https://doi.org/10.1146/annurev-pharmtox-121120-012309
Peeri NC, Shrestha N, Siddikur Rahman M et al (2021) The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: what lessons have we learned? Int J Epidemiol 49:717
Ribrag V, Caballero D, Fermé C et al (2013) Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica. https://doi.org/10.3324/haematol.2012.069757
Saraswat J, Singh P, Patel R (2021) A computational approach for the screening of potential antiviral compounds against SARS-CoV-2 protease: ionic liquid vs herbal and natural compounds. J Mol Liq. https://doi.org/10.1016/j.molliq.2021.115298
Schüttelkopf AW, Van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. https://doi.org/10.1107/S0907444904011679
Soto-Matos A, Szyldergemajn S, Extremera S et al (2011) Plitidepsin has a safe cardiac profile: a comprehensive analysis. Mar Drugs. https://doi.org/10.3390/md9061007
Vargas JRN (2020) The COVID-19 pandemic. Rev Fac Med 68:7
Vishvakarma VK, Kumari K, Patel R et al (2015) Theoretical model to investigate the alkyl chain and anion dependent interactions of gemini surfactant with bovine serum albumin. Spectr Acta Part A Mol Biomol Spectrosc. https://doi.org/10.1016/j.saa.2015.01.068
Vishvakarma VK, Singh P, Kumar V et al (2019) Pyrrolothiazolones as potential inhibitors for the nsP2B-nsP3 protease of dengue virus and their mechanism of synthesis. ChemistrySelect. https://doi.org/10.1002/slct.201901119
Vishvakarma VK, Nand B, Kumar V et al (2020) Xanthene based hybrid analogues to inhibit protease of novel corona virus: molecular docking and ADMET studies. Comput Toxicol. https://doi.org/10.1016/j.comtox.2020.100140
White KM, Rosales R, Yildiz S et al (2021) Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science. https://doi.org/10.1126/science.abf4058
Wu YC, Chen CS, Chan YJ (2020) The outbreak of COVID-19: an overview. J Chinese Med Assoc 83:217
Acknowledgements
The corresponding author, Prashant Singh dedicates this work in the memory his lovely teacher, late Dr. Vandana Uberoi, Associate Professor of Chemistry, Acharya Narendra Dev College, University of Delhi, Delhi, India
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
“I, Prashant Singh (the Corresponding Author), declare that this manuscript is original, has not been published before, and is not currently being considered for publication elsewhere. I further confirm that the order of authors listed in the manuscript has been approved by all of us. The authors also declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Research involving human participants and/or animals
It is declared that no human participants and/or animals are used in this work.
Additional information
Handling editor: F. Albericio.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vishvakarma, V.K., Singh, M.B., Jain, P. et al. Hunting the main protease of SARS-CoV-2 by plitidepsin: Molecular docking and temperature-dependent molecular dynamics simulations. Amino Acids 54, 205–213 (2022). https://doi.org/10.1007/s00726-021-03098-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-021-03098-1