
Abstract
Transient receptor potential A1 (TRPA1) is implicated in somatosensory processing and pathological pain sensation. Although not strictly voltage-gated, ionic currents of TRPA1 typically rectify outwardly, indicating channel activation at depolarized membrane potentials. However, some reports also showed TRPA1 inactivation at high positive potentials, implicating voltage-dependent inactivation. Here we report a conserved leucine residue, L906, in the putative pore helix, which strongly impacts the voltage dependency of TRPA1. Mutation of the leucine to cysteine (L906C) converted the channel from outward to inward rectification independent of divalent cations and irrespective to stimulation by allyl isothiocyanate. The mutant, but not the wild-type channel, displayed exclusively voltage-dependent inactivation at positive potentials. The L906C mutation also exhibited reduced sensitivity to inhibition by TRPA1 blockers, HC030031 and ruthenium red. Further mutagenesis of the leucine to all natural amino acids individually revealed that most substitutions at L906 (15/19) resulted in inward rectification, with exceptions of three amino acids that dramatically reduced channel activity and one, methionine, which mimicked the wild-type channel. Our data are plausibly explained by a bimodal gating model involving both voltage-dependent activation and inactivation of TRPA1. We propose that the key pore helix residue, L906, plays an essential role in responding to the voltage-dependent gating.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transient receptor potential A1 (TRPA1) is a nonselective cation channel highly expressed in a subpopulation of primary afferent sensory neurons of the dorsal root and trigeminal ganglia [16, 42] and implicated in somatosensory processing and pathological pain sensation, particularly inflammatory and neuropathic pain [36]. TRPA1 is activated by a plethora of natural and synthetic compounds, including both electrophilic chemicals and oxidants that covalently modify cysteine residues at the cytoplasmic N-terminus and nonelectrophilic agents that bind to the channel in noncovalent fashions [8, 47]. In addition, the channel is sensitive to intracellular Ca2+ and pH [12, 29, 44, 47, 60], as well as membrane depolarization [29]. The ability of TPRA1 to respond to multiple stimuli is consistent with its role in sensing pain stimuli as well as irritants and allergens [4, 7, 20, 25, 26, 32, 36, 42, 46].
Similar to other TRP channels, e.g., TRPV1 and TRPM8 [48], TRPA1 displays voltage dependence, showing marked outward rectification, especially under weakly activated conditions, such as activation by low temperature, CO2, O2, or intracellular Ca2+ [42, 60]. The rectification becomes less pronounced as the channel is activated strongly by certain chemical ligands [19, 20] or stimulated for a long time period by electrophilic compounds [51], indicating a shift of voltage dependence to more negative potentials. In general, the commonly used TRPA1 agonists, e.g., allyl isothiocyanate (AITC) and cinnamaldehyde, elicit currents with variable degrees of outward rectification depending on the agonist concentration and stimulation duration [51]. Ironically, however, some studies also showed leveling off or inactivation at high positive potentials (e.g., at > +50 mV) for mouse and human TRPA1 expressed in CHO and HEK293 cells [1, 3, 22–24, 35, 36, 45], giving rise to an inwardly rectifying appearance in the current–voltage (I–V) relationships. It was not made clear under which conditions the inward rectification tended to occur and often both linear and inwardly rectifying I–V curves were displayed in the same study. Based on single channel measurements from cell-attached patches, the open probability of TRPA1 clearly shows inactivation at positive potentials [35]. Therefore, it appears that TRPA1 has both voltage-dependent activation and inactivation.
Like other TRP channels, TRPA1 may have the same architecture as voltage-gated, Shaker-type K+ channels, for which two molecular gates exist. The inner gate is formed by bundle crossing of the four S6 transmembrane segments near the cytoplasmic side, while the outer gate involves the selectivity filter situated in the pore loop between the S5 and S6 transmembrane segments [6, 28]. Mutational analyses at the pore loops of TRPV1 [31, 39] and TRPA1 [9] revealed that residues adjacent to the selectivity filter are important for TRP channel gating, suggesting a significant contribution of the outer gate in TRP channel activation. Here, we report an unexpected finding involving L906 in the pore helix of TRPA1. When substituted by another amino acid, including cysteine and 14 others, the resultant channel displays only inward rectification, showing stronger activity at negative than at positive potentials. Such an effect was unaffected by divalent cations. Our results suggest a strong influence of pore helix in voltage-dependent gating of TRPA1.
Materials and methods
cDNA and mutagenesis
The mouse TRPA1 cDNA was a gift from Dr. Gina Story (Washington University in St. Louis). Point mutations were introduced using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the standard PCR overlap extension technique. The mutations were verified by DNA sequencing.
Cell culture and transfection
HEK293 cells were grown in DMEM containing 10 % (vol/vol) fetal bovine serum (FBS), 2 mM l-glutamine at 37 °C in a humidity-controlled incubator with 5 % CO2. All cell culture reagents were purchased from Invitrogen. The conditions for transient transfection of cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in serum-free conditions were optimized. The medium was exchanged for FBS-containing DMEM 6 h after transfection. Transfection efficiency was monitored through cotransfection with an EGFP vector, coding for the enhanced green fluorescent protein. Electrophysiological recordings were performed between 24 and 36 h after transfection.
Electrophysiology
Whole-cell patch-clamp experiments were performed at room temperature (22–24 °C) using an EPC-9 or an EPC-10 amplifier and the PatchMaster software (HEKA). Patch pipettes had a resistance of 2–4 MΩ. Series resistance was compensated at 60–80 %. The normal internal solution consisted of 140 mM CsCl, 10 mM HEPES, 5 mM EGTA, 0.1 mM CaCl2, and 1 mM MgCl2, with pH adjusted to 7.2 by CsOH. The free Ca2+ concentration was ~13–14 nM based on the calculation using Theo’s Chelator program (http://maxchelator.stanford.edu/CaEGTA-TS.htm). The divalent cation-free internal solution contained 140 mM CsCl, 10 mM HEPES, and 10 mM BAPTA, with pH adjusted to 7.2 by CsOH. The standard or physiologically relevant external solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES, with pH adjusted to 7.4 by NaOH. For the Ca2+-free external solution, the 2-mM CaCl2 was replaced by 0.5 mM EGTA in the standard external solution. For the divalent cation-free solution, MgCl2 was omitted from the Ca2+-free external solution. The N-methyl-d-glucamine (NMDG+) solution contained 150 mM NMDG+, 10 mM HEPES, and 5.5 mM glucose, with pH adjusted to 7.4 by HCl. The Na+-only solution contained 150 mM NaCl, 10 mM HEPES, and 5.5 mM glucose, with pH adjusted to 7.4 by NaOH. The osmolarities of external solutions were adjusted to 300 mOsm with sucrose. Except the results shown in Figs. 1 and 2, all recordings were performed using the Ca2+-free external solution unless otherwise stated. Solutions were switched using a gravity-fed continuous focal perfusion system. AITC was used at 100 μM for all experiments. Unless stated otherwise, TRPA1 currents were elicited by applying voltage ramps every 2 s from −100 to +100 mV from the holding potential of 0 or +30 mV over a period of 300 ms, with a 20-ms step at −100 mV before and a 20-ms step at +100 mV after the ramp.

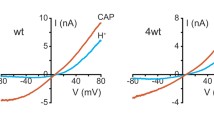
Mutation of L906 at the pore helix of mouse TRPA1 to cysteine converts the channel from outward to inward rectification. a Sequence alignment of putative pore regions of mammalian TRPA1 and TRPM channels. Equivalent regions of known channel structures (KvAP, Kv1.2, KcsA, NaKbc, NaChBac, NavAb, and NavRH) are shown for comparison. Shaded areas indicate minimal spans of pore helices and selectivity filters, adapted for TRPMs from Refs. [27, 34]. Residues mutated in mouse TRPA1 in the current study (Pro904, Leu905, and Leu906) and the Asp (D918) previously shown to determine the Ca2+ selectivity of TRPA1 [51] are underlined. b Whole-cell current recording of a HEK293 cell expressing wild-type mouse TRPA1 in the normal extracellular solution containing 2 mM Ca2+ and 1 mM Mg2+. A 300-ms ramp from −100 to 100 mV was applied every 2 s, from a holding potential of 0 mV and current measured at −100 mV (filled circles) and +100 mV (open circles) during each ramp is plotted as a function of time. AITC (100 μM) was applied during the time indicated by the horizontal bar. The graph below the current traces shows changes in rectification ratio ([R = I −100/I +100] for the same cell during the same time period. The bottom plot shows current–voltage (I–V) relationships recorded from the same cell by the voltage ramp collected at basal (black line), the peak of AITC response (red line), and the end of time course (blue line). c As in b, but for a cell that expressed L906C mutant of mouse TRPA1. Note the inwardly rectifying I–V relationships and the large R values

Inward rectification of L906C is independent of divalent cations. Similar to Fig. 1b, c, but the recording was performed using a Ca2+-free (omitting Ca2+ and adding 0.5 mM EGTA) bath solution (a, b) or divalent cation-free bath (omitting Ca2+ and Mg2+ and adding 0.5 mM EGTA) and pipette (omitting Ca2+ and Mg2+ and adding 10 mM BAPTA) solutions (c, d). Representative traces are shown as for Fig. 1b, c for cells that expressed wild-type mouse TRPA1 (a, c) or its L906C mutant (b, d)
For estimation of NMDG+ permeability, the cell was exposed sequentially to NMDG+ and Na+-only solutions, while currents were recorded using the voltage ramp protocol. P NMDG/P Na was determined as the exponential function for the difference between reversal potentials of NMDG+ and Na+ multiplied by F/RT, where F, R, and T have their usual meanings.
Simulation of voltage-dependent activation and inactivation
The conductance–voltage (G–V) relationships for voltage-dependent activation and inactivation were estimated using the Boltzmann sigmoidal equation: G = G min + (G max − G min) / (1 + exp(−(V − V 1/2) / s)), in which G max and G min for the maximum and minimum conductance were set to be 1 and 0, respectively; V 1/2, representing the voltage for reaching 50 % of maximum conductance, was set to a desired value for activation or inactivation; s (=RT/zF), representing the slope factors, were set to be 47 for activation based on the mean value estimated from previous publications [24, 44, 60] and −20 for inactivation according to the current study. The voltage-dependent conductance for a given potential was estimated as G (v) = G act × G inact. For a more realist prediction of I–V relationships, we included 5 % equivalent of the G max as voltage-independent conductance to account for leak and voltage-independent TRPA1 activities. Therefore, the total conductance at a given potential was G total = 0.05 + 0.95G (v) and the relative current was determined as I/I max = (V − V rev) × G total, where the reversal potential V rev, was set to 0 for TRPA1.
Chemicals
AITC, HC-030031, menthol, and ruthenium red (RR) were purchased from Sigma-Aldrich Co.
Statistical analysis
Data are presented as means ± SEM. Statistical significance was assessed using Student’s t test unless otherwise stated. For all results, asterisks indicate the following: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Mutating L906 converts TRPA1 into an inwardly rectifying channel
Because of the limited sequence homology, the published alignments between TRPA1 and channels of known structures, e.g., KcsA, at the pore region differ between studies [51, 53]. We have adopted the alignment used by **ao et al. [53] and added the equivalent regions of several other K+ channels, nonselective cation channels, and the recently resolved bacterial Na+ channels [57], as well as that predicted for TRPM channels [27, 34], to the alignment in order to define the minimal motifs for pore helix and selectivity filter of TRPA1 (Fig. 1a). Because aspartic acid 918 (D918) of rat TRPA1 determines Ca2+ permeability and is included in the selectivity filter [51], it is also reasonable to assume that residues preceding D918 constitute the selectivity filter and pore helix of TRPA1. We focused on the proline–leucine–leucine (PLL) motif because it represents the start of an alpha helix according to secondary structure prediction, and in some Kv channels, the PAG or PVP motif in the S6 segment has been proposed to form a “hinge” that contributes to channel gating [15, 40, 52, 57]. To test the role of the PLL motif in TRPA1 channel function, we changed PLL individually into a cysteine by site-directed mutagenesis. We then expressed the mutant construct in HEK293 cells by transient transfection and examined ionic currents under basal and AITC-stimulated conditions using whole-cell recordings.
Cells expressing wild-type mouse TRPA1 displayed minimal current under the basal nonstimulated condition in the physiological external solution containing 2 mM Ca2+ and 1 mM Mg2+. Perfusion with AITC (100 μM) produced an increase in membrane currents, which was slow initially but became robust after 10–20 s. This fast phase of current development was followed by rapid decreases of currents at both positive and negative potentials, indicating desensitization (Fig. 1b). The complex kinetics of AITC-induced current changes represent slow action of AITC as an agonist and a strong potentiation, followed by inhibition, by Ca2+ that entered the cell through the activated TRPA1 channels [3, 5, 36, 51]. Under these conditions as outlined, the currents were outwardly rectifying (Fig. 1b), with the rectification ratio (R), calculated as the ratio of absolute current amplitude at −100 mV over that at +100 mV, maintained at less than 1 (0.72 ± 0.07, n = 12). Here, we use R < 1 to indicate outward rectification and R > 1 for inward rectification. The values are also informative for relative rectifications at outward and inward directions. For the three mutations at PLL, P904C displayed similar phenotype as the wild-type channel. L905C had a reduced and slower response to AITC stimulation, but the rectification ratio did not differ from the wild-type channel. Surprisingly, L906C displayed constitutive currents, which were larger in the negative potentials than in the positive ones, with an average R value of 2.93 ± 0.43 (n = 13). Stimulation by AITC (100 μM) further enhanced the currents at both negative and positive potentials, with kinetic profiles resembling that of the wild-type channel, i.e., activation followed by desensitization (Fig. 1c). The R value at the peak of AITC-stimulated currents was 3.16 ± 0.40 (n = 13) (Fig. 1c). The I–V curves for L906C obtained by voltage ramps from −100 to +100 mV showed inward rectification with current decreased at high positive potentials and a reversal potential near 0 (1.0 ± 1.3 mV, n = 13), which was not different from that of wild-type TRPA1 (0.8 ± 1.4, n = 12, p = 0.9 vs L906C by unpaired t test). Despite the large change in the rectification ratio, the current densities at −100 mV at the peak of AITC stimulation were not significantly different between wild type (−0.59 ± 0.10 nA/pF, n = 12) and L906C (−0.36 ± 0.08 nA/pF, n = 13, p = 0.084 vs wild type by unpaired t test). We therefore focused on mutations at L906 in subsequent studies.
The inward rectification of L906C is not dependent on divalent cations
Pore block by divalent cations is a common cause of rectification for many ion channels [33], including some TRP channels [49, 54]. Although outward rectification of the wild-type TRPA1 channel is not dependent on the presence of divalent cations in the extracellular solution [51], Ca2+ and/or Mg2+, which are typically found in physiological solutions, could in some way contribute to the rectification behavior of the channel when they are present in the intracellular and/or extracellular solutions. We therefore examined whether divalent cations were responsible for the inward rectification of the L906C TRPA1 mutant. First, we used a Ca2+-free external solution (omitting Ca2+ and adding 0.5 mM EGTA) and found that removal of extracellular Ca2+ markedly slowed the activation and desensitization kinetics without changing the overall current densities (−0.45 ± 0.17 nA/pF for wild type and −0.58 ± 0.09 nA/pF for L906C at −100 mV, n = 7 for each) and rectification behaviors (R = 0.40 ± 0.02 for wild type and 8.42 ± 0.58 for L906C, n = 7 for each) of wild-type TRPA1 and its L906C mutant (Fig. 2a, b). We then used divalent cation-free extracellular (omitting Ca2+ and Mg2+ and including 0.5 mM EGTA) and intracellular (omitting Ca2+ and Mg2+ and including 10 mM BAPTA) solutions. For the wild-type channel, removal of divalent cations resulted in the loss of rectification (R = 1.05 ± 0.01, n = 5, p < 0.05 vs the R value for wild-type TRPA1 in normal physiological solutions) at the peak of the response to AITC. Consistent with the previous report [51] and with the response in the Ca2+-free external solution, the currents developed slowly in response to AITC and strong outward rectification was seen during the first minute of the agonist stimulation (Fig. 2c). Noticeably, under both conditions, the outward currents developed faster and also entered desensitization earlier than the inward currents. The overall rates of current development and desensitization in the external Ca2+-free and divalent cation-free conditions were slower than in the presence of Ca2+ and Mg2+, indicating that although divalent cations, especially Ca2+, are not required for TRPA1 current development and desensitization, they strongly influence the kinetics of these processes.
On the other hand, removal of divalent cations from both bath and pipette solutions did not significantly alter inward rectification of the L906C mutant channel under basal and AITC-stimulated conditions (R = 3.1 ± 2.3 and 3.7 ± 1.5 at basal and the peak of AITC stimulation, respectively) (Fig. 2d). Therefore, the inward rectification of the L906C mutant was not due to a change in the pore block by divalent cations. Similar to wild-type TRPA1, the kinetics of activation and desensitization of the L906C mutant in response to AITC stimulation were also markedly slowed under the divalent cation-free conditions (Fig. 2d), suggesting that Ca2+ and/or Mg2+ could still facilitate the activation of L906C by AITC and the resultant desensitization. In the divalent cation-free solutions, the current densities at −100 mV at the peak of AITC stimulation, −2.03 ± 0.15 nA/pF for wild type (n = 5) and −1.93 ± 0.19 nA/pF for L906C (n = 4), were higher than those in normal physiological solutions or in the Ca2+-free external solution, which could result from removal of the inhibitory effect of Mg2+ on TRPA1 currents [51]. Together, these observations are consistent with the idea that divalent cations are generally inhibitory to TRPA1 function despite initial facilitation. Furthermore, the cysteine substitution at L906 did not alter the effect of divalent cations on channel function. Because the Ca2+-free external solution slowed desensitization without affecting current density, we used it as the bath solution for all subsequent experiments.
L906C is inactivated at positive potentials in a time- and voltage-dependent manner
The representative I–V trace for L906C obtained from the voltage ramp protocol shows a gradual decline of the outward currents after reaching the peak (Fig. 1c), indicative of a voltage- and time-dependent “inactivation.” To examine this possibility, we recorded the basal and AITC-stimulated currents of L906C at −100, −60, and +60 mV using voltage steps from the holding potential of 0 mV. As shown in Fig. 3, currents developed rapidly at all potentials and were increased in response to AITC with activation and desensitization kinetics similar to those obtained from the voltage ramps. However, while the current at −100 and −60 mV remained relatively consistent during the 500-ms steps, that at +60 mV declined exponentially and rapidly such that the steady-state current at the end of the 500-ms step approached 0 (Fig. 3c). The time-dependent inactivation at +60 mV occurred in the absence or presence of AITC and during desensitization; therefore it was independent of the stimulation status of the channel.

Time-dependent inactivation of L906C at positive potentials. Representative current traces of TRPA1 L906C mutant expressed in HEK293 cells recorded by voltage-step protocols. The cell was held at 0 mV and stepped to −60 mV (a), −100 mV (b), or +60 mV (c) for 500 ms at 5 s intervals. Left panels show current traces before (basal, dashed lines), at the peak of (peak AITC, solid black lines), and at the end of (desensitized, solid gray lines) AITC stimulation. Right panels show time courses of currents at the beginning (instantaneous, open circles) and the end (steady-state, filled circles) of the voltage steps during AITC stimulation. Before the step protocols, the standard voltage ramp protocol was applied to test whether the cell could respond to AITC. Thus, the starting point of the time course was not time 0. Similar phenomena were observed in more than three cells for each experiment
To further explore the voltage dependence of the L906C mutant, we applied a series of voltage steps from −100 to +100 mV with a 20-mV increment from the holding potential of 0 mV, followed by a hyperpolarization step to −100 mV (inset in Fig. 4a) to cells that expressed either the wild-type mouse TRPA1 or its L906C mutant. Consistent with previous studies [29], wild-type TRPA1 was only activated at high positive potentials in the absence of an agonist (Fig. 4a). In contrast, the L906C mutant displayed nearly steady currents during the 800-ms voltage steps at all negative potentials. Only a small decrease in current was observed at hyperpolarized negative potentials (<20 % at −100 mV). In contrast, all currents at positive potentials showed rapid inactivation (Fig. 4b). Plotting the time constants of inactivation at +20, +40, +60, +80, and +100 mV against voltages revealed a clear trend of voltage-dependent acceleration of inactivation for L906C (Fig. 4c). The I–V curves generated from the steady-state currents at the end of the 800-ms voltage steps clearly show that the mutation at L906 switched the polarity of voltage dependence of TRPA1, converting the channel from outwardly to inwardly rectifying (Fig. 4d). This change was caused, apparently, by constitutive opening of the mutant channel at negative potentials and voltage-dependent inactivation at positive potentials (Figs. 3c and 4c). Interestingly, while the inactivation of L906C at positive potentials was time-dependent, the “re-opening” was very rapid upon step** to the hyperpolarized potential (−100 mV). Tail current traces from all positive potentials nearly overlapped (Fig. 4b), indicating a very quick recovery from inactivation.

Voltage-dependent inactivation of L906C. a, b Representative current traces induced by the step protocol indicated in the inset from HEK293 cells expressing wild-type TRPA1 (a) and its L906C mutant (b) under basal unstimulated conditions. c Histogram showing time constants of inactivation of outward currents of L906C at indicated step potentials. The time constant was calculated by fitting the currents with a single-exponential equation. Data are means ± SEM for three cells. d I–V relationships based on the steady-state currents at the end of the voltage steps for wild-type and mutant TRPA1 channels. Data are means ± SEM for three cells. e Conductance–voltage (G–V) relationships for L906C under basal and AITC-stimulated conditions. To correct for the changing activity levels during AITC stimulation, a 50-ms pre-step to −40 mV was included before the test step (inset at bottom right) and the current amplitude at the end of the pre-step was used to normalize the tail current measured at +100 mV. Representative traces are shown to the right of the G–V curves, which were fitted by the Boltzmann sigmoidal equation, with V 1/2 values indicated
To quantify the voltage dependence of the mutant channel, we applied a series of voltage steps from −100 to +100 mV with a 10-mV increment from the holding potential of +30 mV, followed by a step to +100 mV, to cells that expressed L906C under basal and AITC-stimulated conditions (Fig. 4e). Because of the time-dependent changes during activation and desensitization induced by AITC (for example, see Fig. 3), the activity levels were not stable throughout the time period needed to complete all steps (~20 s). Therefore, to calibrate the relative activity level for each step, a 50-ms pre-step to −40 mV was included before the test step (see voltage protocol in the inset of Fig. 4e), and the current amplitude at the end of the pre-step was used to normalize the tail current measured at +100 mV before the relative conductance (normalized I tail or G/G max) was calculated and plotted against the step voltage. Fitting the conductance–voltage (G–V) plots with the Boltzmann equation yielded half voltage for inactivation (V 1/2) of −2.6 ± 1.6 mV (n = 12) and 9.8 ± 1.6 mV (n = 14) for basal and AITC-stimulated conditions, respectively (Fig. 4e). These values are lower than that reported for wild-type TRPA1 (34.5 mV) based on single channel open probability recorded from cell-attached patches [35]. The negative slopes of the G–V curves (slope factors of −21.9 ± 1.2 for basal and −18.4 ± 1.1 for AITC-stimulated) are consistent with the mutant channel being activated by hyperpolarization rather than depolarization.
L906C mutation results in additional property changes
AITC is an electrophilic compound that activates TRPA1 through covalent modification of cysteine residues at the cytoplasmic N-terminus [8, 47]. Since L906C is constitutively active, it is unlikely that the inward rectification resulted from interaction between AITC and the introduced cysteine residue. Nonetheless, because TRPA1 is also activated by some nonelectrophilic compounds, such as menthol [22], we tested the effect of menthol on L906C. To our surprise, although menthol (200 μM) activated wild-type TRPA1 with outwardly rectifying I–V relationships, it failed to change the activity of L906C (Fig. 5a, b, d). At higher concentrations, menthol also inhibits TRPA1 [22]. Interestingly, despite the lack of stimulatory effect at 200 μM, 1 mM menthol significantly inhibited the basal constitutive current of L906C (Fig. 5c, d).

Effect of menthol on L906C. a, b Representative I–V traces (left) and time courses at +100 and −100 mV (right) of whole-cell currents obtained by voltage ramps from cells that expressed L906C mutant (a) and the wild-type (b) constructs of TRPA1. Menthol (men, 0.2 mM) was applied as indicated. I–V traces show basal and during menthol application. c Similar to a, but 1 mM menthol was applied to the cell that expressed L906C, which caused inhibition. d Summary data (means ± SEM, n = 6) for experiments shown in a–c. *p < 0.05, **p < 0.01, ***p < 0.001 vs basal by paired t test
Another property of TRPA1 is that it undergoes pore dilation in response to agonist stimulation [10]. We examined this property by comparing the permeability to NMDG+ with that to Na+. As determined using reversal potential values obtained in isotonic NMDG+ and Na+ extracellular solutions, for wild-type TRPA1, P NMDG/P Na ratio changed from 0.15 ± 0.01 (n = 7) immediately following AITC (100 μM) application to 0.27 ± 0.03 (n = 7) at 3 min after AITC treatment. However, the P NMDG/P Na ratio (0.16 ± 0.01 before and 0.18 ± 0.01 at 3 min after AITC application) was not significantly changed for L906C. This result suggests that unlike the wild-type channel, the L906C mutant of TRPA1 does not undergo pore dilation in response to AITC stimulation.
We also tested the response of L906C to TRPA1 blockers. L906 is located in the pore helix of TRPA1 close to D918 (Fig. 1a), which is considered a part of the selectivity filter and critical for Ca2+ permeation [51]. Consistent with its pore localization, the mutation at L906 markedly reduced sensitivity of the channel to inhibition by the nonspecific pore blocker, ruthenium red (RR), at negative potentials (Fig. 6a–c). The IC50 of RR for current at −100 mV changed from 0.76 ± 0.06 μM for wild-type TRPA1 (n = 5) to 8.06 ± 0.31 μM for L906C (n = 4, p < 0.01 vs wild type). Similar to that of wild-type channel, the outward currents of L906C at positive potentials were not very sensitive to RR (Fig. 6a, b). Interestingly, the L906C mutation also dramatically reduced the inhibitory capability of HC030031, a noncharged TRPA1 selective inhibitor [13]. For the wild-type TRPA1, 30 μM HC030031 almost completely inhibited both the inward and outward currents (Fig. 6d). However, the same concentration of HC030031 only slightly inhibited the currents of L906C (Fig. 6e). Accordingly, the concentration–response curve to HC030031 was right-shifted for L906C as compared to the wild-type channel (Fig. 6f), with the IC50 for currents at −100 mV increased from 10.10 ± 0.07 μM for wild-type TRPA1 (n = 4) to 37.8 ± 0.49 μM for L906C (n = 3, p < 0.05 vs wild type). This decrease in the inhibitory effect of HC030031 could suggest that the site of action of this compound might also be near the pore region; however, further experimentation is needed to test this possibility.

Reduced sensitivity of L906C to inhibition by ruthenium red and HC030031. a–c Inhibitory effects of ruthenium red (RR). a, b Representative I–V traces under basal, AITC (100 μM)-stimulated, and AITC plus RR (10 μM) conditions obtained from voltage ramps for cells that expressed wild-type TRPA1 (a) and its L906C mutant (b). c Concentration–response curves of RR for wild type and L906C at −100 mV. Data are means ± SEM for four to five cells and fitted with the Hill equation. d–f Inhibitory effects of HC030031 (HC). d, e Representative I–V traces under basal, AITC (100 μM)-stimulated, and AITC plus HC (30 μM) conditions obtained from voltage ramps for cells that expressed wild-type TRPA1 (d) and its L906C mutant (e). f Concentration–response curves of HC for wild type and L906C at −100 mV. Data are means ± SEM for three to four cells and fitted with the Hill equation
Side chain properties for residue at position 906 to confer inward rectification
We further examined the side chain properties that confer inward rectification of TRPA1 at residue position 906 by substituting the leucine with all other 18 amino acids. Upon expression in HEK293 cells, the L906P, L906D, and L906E mutants showed very small basal currents, which were only slightly increased by stimulation with 100 μM AITC (Fig. 7a, also see Fig. 8 for representative I–V traces). With the low activity, rectification was not obvious for these mutants, but the currents at −100 mV were largely reduced by ruthenium red (10 μM) or when extracellular Na+ was replaced with NMDG+ (Fig. 8), indicating cation conductance, likely through the expressed mutant channels. For the other 15 substitutions, all but one, L906M, showed inward rectification (Fig. 7a, b). Many of the mutants also displayed high, inwardly rectifying basal activities. Stimulation by AITC (100 μM) elicited quite variable responses with no obvious correlation to the side chain property of the residue. Remarkably, conserved substitutions of the leucine with valine and isoleucine also resulted in inward rectification, with L906V showing high basal activity and moderate further increase in response to AITC while L906I displayed low basal current and robust response to the channel agonist (Fig. 7a, d). Typically, AITC either did not change or increased the rectification ratios of the mutant channels with exception of L906F, which showed a marked decrease in the rectification ratio in response to AITC (p = 0.0049, n = 9, basal vs AITC by paired t test) (Figs. 7a, b and 8). The L906M mutation had relatively low basal activity, but responded to AITC with robust current, which had a nearly linear I–V relationship (R = 0.92 ± 0.05, n = 8) and was blocked by RR (Fig. 7a–c). In addition to the examples shown in Fig. 7c–e, the representative I–V traces for all other tested mutants are shown in Fig. 8. Together, these data demonstrate the unique role and absolute requirement for leucine at position 906 to confer the outwardly rectifying voltage dependence of the TRPA1 channel. Substitutions at this position by most other amino acids render a reversal of the voltage dependence, with few exceptions, e.g., methionine. A few substitutions also severely compromised channel function (aspartate, glutamate, and proline).

Most mutations at L906 convert the TRPA1 channel to inward rectification. a, b Current densities (a) at +100 mV (upper graph) and −100 mV (lower graph) and the rectification ratios (b) under basal (open bars) and AITC-stimulated (100 μM, filled bars) conditions for HEK293 cells expressing wild-type mouse TRPA1 (L) and its mutations with L906 substituted by the indicated amino acids. Dashed line in b indicates R = 1, which separates outward rectification (R < 1) from inward rectification (R > 1). Data are means ± SEM from 4 to 13 cells for each construct. c–e Left panels, representative I–V traces under basal (black lines), AITC-stimulated (100 μM, red lines), and AITC plus ruthenium red (RR, 10 μM, blue lines) conditions for selected L906 substitutions as indicated. Right panels, time courses of currents at −100 and +100 mV for the corresponding mutants shown to the left. AITC and RR were applied as indicated by the horizontal bars

I–V relationships of L906 mutants. Representative I–V traces obtained using voltage ramps from −100 to +100 mV at basal (black), peak of stimulation by AITC (100 μM, red), after addition of ruthenium red (RR, 10 μM, blue), or replacement of cations in the bath solution with NMDG+ (green) for HEK293 cells that expressed L906 mutants of mouse TRPA1 as indicated. Whole-cell recordings were performed using the Ca2+-free bath solution as described in “Materials and methods.” Summary data for current densities and rectification ratios under basal and AITC-stimulated conditions are shown in Fig. 7a, b
Discussion
Bimodal voltage dependency of TRPA1
Although TRPA1 and other thermosensitive TRP channels, such as TRPV1 and TRPM8, are not activated by voltage alone within physiologically relevant potentials, they show voltage dependence [48]. Other physical and chemical stimuli can shift the voltage dependence to the physiological range, leading to the proposal of this being a main mechanism underlying TRP channel activation [48, 58]. Compared to TRPV1 and TRPM8, the voltage-dependent regulation of TRPA1 appears more complex, showing both activation and inactivation at positive voltages by different research groups [1, 3, 5, 23, 29, 36, 44, 51, 60]. It is also not uncommon that linear I–V relationships are reported between −100 and +100 mV when the channel is strongly activated. Outward rectification is commonly observed when activated by certain chemical agonists, low temperature, and intracellular Ca2+ [29, 60].
There are two major mechanisms for voltage dependence (rectification) of TRP channels: (a) voltage-dependent pore block by divalent cations and (b) intrinsic voltage-dependent gating [34]. For TRPA1, strong outward rectification was observed in bath solutions that contained neither Ca2+ nor Mg2+ [51] and likewise inactivation at positive potentials was also shown in Ca2+-free external solutions [3, 36], suggesting that the contribution of divalent cations to TRPA1 voltage dependence was minimum. However, because of the complex effect of entering Ca2+ on channel activity [32, 51], divalent cations can affect the apparent rectification via modulation of intrinsic voltage dependence. In this study, we intentionally prolonged the duration of AITC application to allow full current development and its conversion to desensitization. In the presence of Ca2+/Mg2+, AITC induced a biphasic development of TRPA1 currents, with an initial slow phase followed by a rapid increase, which then entered a rapid desensitization in the continued presence of AITC (Fig. 1b). This complex behavior can be explained by Ca2+-dependent potentiation and then inactivation, both involving Ca2+ entry through the open TRPA1 channels [51]. Supporting this idea, removal of extracellular Ca2+ alone slowed both activation and desensitization without affecting maximal current and rectification ratio (Fig. 2a). This would be consistent with the slow and cumulative nature of TRPA1 activation through covalent cysteine modification by the electrophilic AITC [17]. The rectification ratio increased rapidly with Ca2+ but slowly without Ca2+, indicative of negative shifts of the voltage dependence by agonist stimulation. Interestingly, under both conditions, the rectification ratio remained nearly constant during desensitization (Figs. 1b and 2a), indicating that the desensitization results mainly from proportional closure (or removal) of activated channels rather than returning them to a prestimulated state. Removal of divalent cations allowed for more complete, but slower, activation by AITC and eventual loss of outward rectification (Fig. 2c). However, outward rectification (R < 1) was clearly evident during the extended initial activation phase despite the complete lack of Ca2+ and Mg2+ (Fig. 2c). Together, these data confirm that divalent cations are not required for outward rectification of TRPA1, and the loss of rectification and enhanced current amplitude in divalent-free solutions are results of a more negative shift of voltage dependence in the absence of Ca2+/Mg2+-dependent inhibition. Therefore, intrinsic voltage dependence may be the main mechanism underlying outward rectification of TRPA1 currents.
Mutations at L906 convert TRPA1 into inward rectification independent of divalent cations
Interestingly, although the wild-type mouse TRPA1 only displayed outwardly rectifying or linear currents under our experimental conditions, the majority of mutations at L906, located in the pore helix of TRPA1, showed exclusively inwardly rectifying currents with inactivation at positive potentials. This unexpected finding revealed the importance of TRPA1 pore helix in channel gating. The pore loop of TRPA1 has been suggested to contribute to channel gating based on the finding that A946S and M949I substitutions in the upper portion of S6 segment of rat TRPA1 converted the effect of electrophilic thioaminal-containing compounds from activation to inhibition [9]. T633 in TRPV1 pore helix has also been implicated in channel gating by protons [39] and camphor [30]. Mutations in F640 and T641 of TRPV1 also led to constitutive activation and loss of proton response [31]. Interestingly, TRPV1 T633 is in the equivalent position as TRPA1 L906 (Fig. 9a), but mutations at TRPA1 L906 caused more dramatic changes in channel gating. Importantly, only leucine or methionine at this position gives rise to outward rectification or linear currents; 15 other amino acids caused TRPA1 to rectify only inwardly; the remaining three had very low activity. Therefore, L906 is absolutely required for normal voltage-dependent gating of TRPA1. Consistently, the leucine is conserved in nearly all vertebrate TRPA proteins, with the exception of phenylalanine in the frog Xenopus tropicalis (Fig. 9b). The L906F substitution is also unique among others with its markedly reduced rectification ratio in response to AITC (Figs. 7b and 8). In insects and nematodes, the position contains a methionine (Fig. 9b), the only other amino acid in our study that permitted large agonist-induced response without inward rectification.

TRPA1 pore helix and selectivity filter. a Alignment of mouse TRPA1 and TRPVs at putative pore helices (gray) and selectivity filters (yellow). L906 of TRPA1 and T633 of TRPV1 are indicated in red. b Alignment of pore helices and selectivity filters for TRPA1 from selected animal species. The conserved PXLS motif in terrestrial vertebrates is highlighted in gray and the phenylalanine in frog and methionines in insects and nematode at the position equivalent to mouse L906 are highlighted in yellow. GenBank accession numbers are indicated in square brackets
Removal of divalent cations had very little impact on inward rectification of L906C, ruling out the possibility that the mutation acquired a gain-of-function unidirectional pore blockade by Ca2+/Mg2+. Similar to wild-type TRPA1, removal of divalent cations increased the AITC-evoked peak currents of L906C at −100 mV, indicating that Ca2+/Mg2+ inhibition was unaffected by the mutation. It was recently shown that a cluster of acidic residues in the distal C-terminus of TRPA1 may be critical for the Ca2+-induced responses [44]. Since L906 is 12 residues upstream from D918, which determines the Ca2+ selectivity and is in the permeation pathway [51], L906 is likely situated outside of the selectivity filter and thus unlikely involved in pore block by divalent cations.
Voltage-dependent inactivation of L906C
The L906 mutations reveal an interesting feature of intrinsic voltage dependence of TRPA1. Previously, residues in the S6 segment of human TRPA1 have been shown to influence channel rectification [5]. While some of the mutations displayed strong outward rectification without or with little inward currents, G958R, located near the C-terminal end of S6 segment close to the putative helix bundle, showed strong inwardly rectifying constitutive activity with weak response to AITC (200 μM). However, G958R differs from L906C in that the outward currents at positive potentials showed little inactivation during voltage steps. The ~ −13-mV shift in reversal potential of G958R also suggests a change in permeability [5]. Therefore, the inward rectification of G958R likely resulted from permeation block.
By contrast, the L906 mutations represent a change in intrinsic voltage dependence. We show clearly that L906C is inactivated at positive potentials in a time-dependent manner and the time constant of inactivation is dependent on voltage. The G–V curve for L906C has a negative slope, similar to hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. AITC stimulation shifted the G–V curve by about ~12 mV to more positive, but the V 1/2 remains more negative than that for the wild-type TRPA1 determined from cell-attached single channel recordings [35].
Bimodal voltage-dependent gating model for TRPA1
Our data provide a clear demonstration of voltage-dependent inactivation of TRPA1 and suggest an interesting possibility that may explain the bimodal voltage dependence of TRPA1 reported by different research groups. Assuming that both voltage-dependent activation and inactivation are involved in TRPA1 gating, then one should be able to predict the different rectification patterns (outward and inward rectifications as well as linear) by changing the V 1/2 values for activation and inactivation as illustrated in Fig. 10. In this simulation, the V 1/2 for inactivation is assumed to vary greatly under different experimental conditions. This is possible because large voltage shifts in response to stimuli (or modulators) are predicted by the thermodynamic principle from the small gating charge of TRP channels [34]. When V 1/2 for inactivation is high (≥+150 mV), its contribution to the I–V between −100 and +100 mV is negligible and the voltage dependence follows the same principle that has been well described for other TRP channels, i.e., changing from outward rectification to linear as V 1/2 for activation shifts to more negative [34, 48]. Conceivably, factors in recording conditions under certain experimental settings could also negatively shift the inactivation voltage such that voltage-dependent inactivation becomes detectable within the commonly applied potential range (e.g., Fig. 10c). Thus, our model offers a plausible explanation for the bimodal voltage dependence of TRPA1. More importantly, the inward rectification behaviors of TRPA1 L906 mutants can be readily explained using the measured inactivation V 1/2 value, with the assumption that activation V 1/2 is also largely shifted to very negative voltages (e.g., <−150 mV, see Fig. 10d).

Simulation of I–V relationships of TRPA1 using the bimodal voltage-dependent gating model. Left plots show G–V curves for activation (solid blue) and inactivation (dot-dash red) using V 1/2 values as indicated and other parameters described in “Materials and methods.” Right plots show I–V curves between −100 and +100 mV predicted from the G–V relationships at the left side. a–c For wild-type TRPA1, outward rectification (a), linear I–V (b), and inward rectification (c) can be generated by separately changing the V 1/2 values for activation and inactivation without altering any other parameter. d For L906C, the characteristic inwardly rectifying I–V relationship can be predicted using the measured inactivation V 1/2 values for basal (dashed blue) and AITC stimulated (solid red) conditions from Fig. 4e. The activation V 1/2 is assumed to be very negative (<−150 mV). However, to account for the increased current density in response to AITC, channel availability needs to be doubled (solid black)
Our data thus suggest that the pore helix plays a critical role in voltage-dependent gating of TRPA1. The L906C substitution shifted both activation and inactivation voltages to more negative, leading to constitutive activation at negative and inactivation at positive potentials. Interestingly, L906C is constitutively active and the currents can be further enhanced by AITC but not menthol. While AITC caused ~12 mV positive shift of the inactivation voltage, such a change cannot explain the doubled current density of L906C by AITC (Fig. 10d), nor can any change of the activation voltage account for such an increase (simulation data not shown). It is thus likely that in addition to altering the voltage dependence of TRPA1, AITC also increases channel availability. About twice the number of the channel would have to become available in order to simulate the effect of AITC on L906C (Fig. 10d). On the other hand, menthol may activate TRPA1 only through negatively shifting the activation voltage. This idea is consistent with the previous observation that menthol dramatically shifted the activation V 1/2 of TRPA1 from 121 to −285 mV [22]. Such an action would not be expected to enhance L906C current, given the already very negative activation V 1/2 under basal conditions. Ironically, the inhibitory effect of menthol was still readily detectable on L906C. This action does not appear to be voltage dependent. Furthermore, pore dilation was not detected for L906C in response to AITC. This could suggest that pore dilation is associated with the change in voltage-dependent activation, a possibility that requires further investigation.
Our model that both voltage-dependent activation and inactivation are involved in TRPA1 gating is reminiscent of similar bimodal voltage dependence of classical voltage-gated channels [14, 18, 41, 43, 50, 59]. A major distinction between TRPA1 and the well-characterized voltage-gated channels with prominent voltage-dependent inactivation seems that for the latter, the inactivation V 1/2 tends to be more negative than activation V 1/2, giving rise to the “window currents,” which are pivotal for the physiological function of these channels [2, 38, 43, 50]. However, for TRPA1, given the very positive activation V 1/2 under resting conditions (121 to 155 mV) [22, 44, 60], the relatively negative inactivation V 1/2 (34.5 mV) [35] should prevent detection of its current at most potentials. It is interesting that menthol dramatically shifted activation V 1/2 of TRPA1 to more negative potentials about 400 mV (to −285 mV) [22] and a rise in intracellular Ca2+ level could also shift it about 150 mV (to −1 mV) [60]. These large changes are expected because of the small gating charge for TRPA1 activation (~0.4e to 0.8e) [24, 44, 60]. The inactivation gating charge for TRPA1 is estimated to be ~ −1.3e based on our results with L906C (Fig. 4) or −2.4e for the wild-type TRPA1 [35]. It would be interesting to compare the effects of different stimuli on both voltage-dependent activation and inactivation of TRPA1.
While molecular determinants for voltage-dependent activation and inactivation can be different [37, 38, 43, 56], the outer mouth of the pore loop has been implicated in voltage-dependent inactivation of HERG K+ channels [21, 41, 59]. Our data suggest that L906 located in the putative pore helix of TRPA1 is critical for both voltage-dependent activation and inactivation. At least for L906C and several other substitutions, voltage-dependent activation was not evident at potentials > −100 mV regardless of the stimulation status. However, the voltage-dependent inactivation was quite prominent in these mutants, suggesting that both voltage-dependent activation and inactivation had been shifted to more negative as compared to wild type. Alternatively, the mutations could be viewed as having lost the voltage-dependent activation. Further investigations are warranted to distinguish these possibilities and to decipher the differences of voltage dependency among various L906 substitutions.
How would mutations at pore helix switch the polarity of response to voltage? It has been suggested for TRPV5 that gating is accompanied with a rotation of the pore helix [55]. Perhaps, voltage sensor movements are translated to pore helix rotations in TRPA1 to subsequently control its gating. The side chain of L906 appears optimal for kee** TRPA1 closed at physiological potentials. Substitutions at this position could place the pore helix at different rotation angles and alter the activation/inactivation responses to voltage sensor movements. In this context, L906 is not necessarily a part of the voltage sensor, but rather a gate keeper. The rotation of the pore helix could affect gating by changing (a) conformation of the selectivity filter and (b) orientation of the S6 segment, which is in close proximity to the pore helix [11] and directly controls the helix bundle. Therefore, either the outer or the inner gate, or both, may be regulated by the pore helix rotation.
Our findings thus suggest that both voltage-dependent activation and inactivation are involved in TRPA1 gating and the pore helix plays a pivotal role in this bimodal channel regulation by voltage. The notion that TRPA1 is under bimodal regulation by voltage should help explain the complexity of TRPA1 gating and I–V relationships observed under different conditions and in different laboratories. Our data that L906 is critical for controlling TRPA1 gating offers important new insights into the structural mechanisms governing pore conformational changes in response to various stimuli, which form the main theme of functional regulation of TRP channels.
References
Alpizar YA, Gees M, Sanchez A, Apetrei A, Voets T, Nilius B, Talavera K (2013) Bimodal effects of cinnamaldehyde and camphor on mouse TRPA1. Pflugers Arch 465:853–864. doi:10.1007/s00424-012-1204-x
Amin AS, Asghari-Roodsari A, Tan HL (2010) Cardiac sodium channelopathies. Pflugers Arch 460:223–237. doi:10.1007/s00424-009-0761-0
Andersson DA, Gentry C, Moss S, Bevan S (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28:2485–2494. doi:10.1523/JNEUROSCI.5369-07.2008
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124:1269–1282
Benedikt J, Samad A, Ettrich R, Teisinger J, Vlachova V (2009) Essential role for the putative S6 inner pore region in the activation gating of the human TRPA1 channel. Biochim Biophys Acta 1793:1279–1288. doi:10.1016/j.bbamcr.2009.04.014
Bernèche S, Roux B (2005) A gate in the selectivity filter of potassium channels. Structure 13:591–600
Bessac BF, Jordt SE (2008) Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 23:360–370. doi:10.1152/physiol.00026
Cavanaugh EJ, Simkin D, Kim D (2008) Activation of transient receptor potential A1 channels by mustard oil, tetrahydrocannabinol and Ca2+ reveals different functional channel states. Neuroscience 154:1467–1476. doi:10.1016/j.neuroscience.2008.04.048
Chen J, Zhang XF, Kort ME, Huth JR, Sun C, Miesbauer LJ, Cassar SC, Neelands T, Scott VE, Moreland RB, Reilly RM, Hajduk PJ, Kym PR, Hutchins CW, Faltynek CR (2008) Molecular determinants of species-specific activation or blockade of TRPA1 channels. J Neurosci 28:5063–5071. doi:10.1523/JNEUROSCI.0047-08.2008
Chen J, Kim D, Bianchi BR, Cavanaugh EJ, Faltynek CR, Kym PR, Reilly RM (2009) Pore dilation occurs in TRPA1 but not in TRPM8 channels. Mol Pain 5:3. doi:10.1186/1744-8069-5-3
Cuello LG, Jogini V, Cortes DM, Perozo E (2010) Structural mechanism of C-type inactivation in K+ channels. Nature 466:203–208. doi:10.1038/nature09153
Doerner JF, Gisselmann G, Hatt H, Wetzel CH (2007) Transient receptor potential channel A1 is directly gated by calcium ions. J Biol Chem 282:13180–13189
Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48. doi:10.1186/1744-8069-4-48
Fox AP (1981) Voltage-dependent inactivation of a calcium channel. Proc Natl Acad Sci U S A 78(2):953–956
Gao Z, Zhang T, Wu M, **ong Q, Sun H, Zhang Y, Zu L, Wang W, Li M (2010) Isoform-specific prolongation of Kv7 (KCNQ) potassium channel opening mediated by new molecular determinants for drug-channel interactions. J Biol Chem 285:28322–28332. doi:10.1074/jbc.M110.116392
Haas ET, Rowland K, Gautam M (2011) Tooth injury increases expression of the cold sensitive TRP channel TRPA1 in trigeminal neurons. Arch Oral Biol 56:1604–1609. doi:10.1016/j.archoralbio.2011.06.014
Hinman A, Chuang HH, Bautista DM, Julius D (2006) TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103:19564–19568
Hodgkin AL, Huxley AF (1952) The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol 116:497–506
Hu H, Tian J, Zhu Y, Wang C, **ao R, Herz JM, Wood JD, Zhu MX (2010) Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch 459:579–592. doi:10.1007/s00424-009-0749-9
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Högestätt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427:260–265
Ju P, Pages G, Riek RP, Chen PC, Torres AM, Bansal PS, Kuyucak S, Kuchel PW, Vandenberg JI (2009) The pore domain outer helix contributes to both activation and inactivation of the HERG K+ channel. J Biol Chem 284:1000–1008. doi:10.1074/jbc.M806400200
Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T, Nilius B (2007) Bimodal action of menthol on the transient receptor potential channel TRPA1. J Neurosci 27:9874–9884
Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T, Nilius B (2008) Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5-biphosphate manipulators. Pflugers Arch 457:77–89. doi:10.1007/s00424-008-0493-6
Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, Voets T (2009) TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci U S A 106:1273–1278. doi:10.1073/pnas.0808487106
Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP (2006) TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50:277–289
Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL (2009) TRPA1 modulates mechanotransduction in cutaneous sensory neurons. J Neurosci 29:4808–4819. doi:10.1523/JNEUROSCI.5380-08.2009
Latorre R, Brauchi S, Orta G, Zaelzer C, Vargas G (2007) ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium 42:427–438
Long SB, Campbell EB, Mackinnon R (2005) Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309:903–908
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445:541–545
Marsakova L, Touska F, Krusek J, Vlachova V (2012) Pore helix domain is critical to camphor sensitivity of transient receptor potential vanilloid 1 channel. Anesthesiology 116:903–917. doi:10.1097/ALN.0b013e318249cf62
Myers BR, Bohlen CJ, Julius D (2008) A yeast genetic screen reveals a critical role for the pore helix domain in TRP channel gating. Neuron 58:362–373. doi:10.1016/j.neuron.2008.04.012
Nagata K, Duggan A, Kumar G, García-Añoveros J (2005) Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci 25:4052–4061
Nichols CG, Lopatin AN (1997) Inward rectifier potassium channels. Annu Rev Physiol 59:171–191
Nilius B, Talavera K, Owsianik G, Prenen J, Droogmans G, Voets T (2005) Gating of TRP channels: a voltage connection? J Physiol 567:35–44
Nilius B, Prenen J, Owsianik G (2011) Irritating channels: the case of TRPA1. J Physiol 589:1543–1549. doi:10.1113/jphysiol.2010.200717
Nilius B, Appendino G, Owsianik G (2012) The transient receptor potential channel TRPA1: from gene to pathophysiology. Pflugers Arch 464:425–458. doi:10.1007/s00424-012-1158-z
Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M (2003) Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci U S A 100:10534–10539
Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M (2005) Regional specificity of human ether-a′-go-go-related gene channel activation and inactivation gating. J Biol Chem 280:7206–7217
Ryu S, Liu B, Yao J, Fu Q, Qin F (2007) Uncoupling proton activation of vanilloid receptor TRPV1. J Neurosci 27:12797–12807
Seebohm G, Strutz-Seebohm N, Ureche ON, Baltaev R, Lampert A, Kornichuk G, Kamiya K, Wuttke TV, Lerche H, Sanguinetti MC, Lang F (2006) Differential roles of S6 domain hinges in the gating of KCNQ potassium channels. Biophys J 90:2235–2244
Smith PL, Baukrowitz T, Yellen G (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379:833–836
Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829
Stotz SC, Jarvis SE, Zamponi GW (2004) Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol 554:263–273
Sura L, Zíma V, Marsakova L, Hynkova A, Barvík I, Vlachova V (2012) C-terminal acidic cluster is involved in Ca2+-induced regulation of human transient receptor potential ankyrin 1 channel. J Biol Chem 287:18067–18077. doi:10.1074/jbc.M112.341859
Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T (2009) Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 12:1293–1299. doi:10.1038/nn.2379
Taylor-Clark TE, Ghatta S, Bettner W, Undem BJ (2009) Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol Pharmacol 75:820–829. doi:10.1124/mol.108.054445
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andrè E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104:13519–13524
Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B (2004) The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430:748–754
Voets T, Janssens A, Prenen J, Droogmans G, Nilius B (2003) Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. J Gen Physiol 121:245–260
Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL (1997) A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol 502:45–60
Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER (2008) The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem 283:32691–32703. doi:10.1074/jbc.M803568200
Webster SM, Del Camino D, Dekker JP, Yellen G (2004) Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 428:864–868
**ao B, Dubin AE, Bursulaya B, Viswanath V, Jegla TJ, Patapoutian A (2008) Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J Neurosci 28:9640–9651. doi:10.1523/JNEUROSCI.2772-08.2008
**ao R, Tang J, Wang C, Colton CK, Tian J, Zhu MX (2008) Calcium plays a central role in the sensitization of TRPV3 channel to repetitive stimulations. J Biol Chem 283:6162–6174. doi:10.1074/jbc.M706535200
Yeh BI, Kim YK, Jabbar W, Huang CL (2005) Conformational changes of pore helix coupled to gating of TRPV5 by protons. EMBO J 24:3224–3234
Zhang M, Liu J, Tseng GN (2004) Gating charges in the activation and inactivation processes of the HERG channel. J Gen Physiol 124:703–718
Zhang X, **a M, Li Y, Liu H, Jiang X, Ren W, Wu J, DeCaen P, Yu F, Huang S, He J, Clapham DE, Yan N, Gong H (2013) Analysis of the selectivity filter of the voltage-gated sodium channel Na(v)Rh. Cell Res 23:409–422. doi:10.1038/cr.2012.173
Zhu MX (2007) Understanding the role of voltage gating of polymodal TRP channels. J Physiol 585:321–322
Zou A, Xu QP, Sanguinetti MC (1998) A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J Physiol 509(Pt 1):129–137
Zurborg S, Yurgionas B, Jira JA, Caspani O, Heppenstall PA (2007) Direct activation of the ion channel TRPA1 by Ca2+. Nat Neurosci 10:277–279
Acknowledgments
We thank Drs. Jun Chen, Haoxing Xu, and Feng Qin for helpful discussions of the project. This work was supported by the National Key Basic Research Program of China (2013CB910604), the National Science and Technology Major Project on “Key New Drug Creation and Manufacturing Program” (2013ZX09103001-016), the National Natural Science Foundation of China Grant for Excellent Key Laboratory (81123004), and the Special Research Foundation of Chinese Academy of Sciences (F030304:61327014). The research in MXZ and ML laboratories are supported in part by grants from the US National Institutes of Health, R01DK081654 and U54MH084691, respectively.
Conflict of interest
The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wan, X., Lu, Y., Chen, X. et al. Bimodal voltage dependence of TRPA1: mutations of a key pore helix residue reveal strong intrinsic voltage-dependent inactivation. Pflugers Arch - Eur J Physiol 466, 1273–1287 (2014). https://doi.org/10.1007/s00424-013-1345-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1345-6