
Abstract
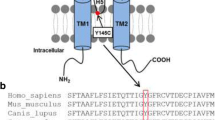
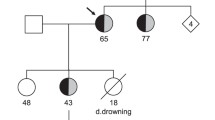
Andersen-Tawil syndrome (ATS) is characterized by dysmorphic features, periodic paralyses and abnormal ventricular repolarization. After genoty** a large set of patients with congenital long-QT syndrome, we identified two novel, heterozygous KCNJ2 mutations (p.N318S, p.W322C) located in the C-terminus of the Kir2.1 subunit. These mutations have a different localization than classical ATS mutations which are mostly located at a potential interaction face with the slide helix or at the interface between the C-termini. Mutation carriers were without the key features of ATS, causing an isolated cardiac phenotype. While the N318S mutants regularly reached the plasma membrane, W322C mutants primarily resided in late endosomes. Co-expression of N318S or W322C with wild-type Kir2.1 reduced current amplitudes only by 20–25 %. This mild loss-of-function for the heteromeric channels resulted from defective channel trafficking (W322C) or gating (N318S). Strikingly, and in contrast to the majority of ATS mutations, neither mutant caused a dominant-negative suppression of wild-type Kir2.1, Kir2.2 and Kir2.3 currents. Thus, a mild reduction of native Kir2.x currents by non dominant-negative mutants may cause ATS with an isolated cardiac phenotype.









Similar content being viewed by others

Abbreviations
- CPVT:
-
Catecholaminergic polymorphic ventricular tachycardia
- EGFP:
-
Enhanced green fluorescent protein
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PMVT:
-
Polymorphic ventricular tachycardia
References
Ai T, Fujiwara Y, Tsuji K, Otani H, Nakano S, Kubo Y, Horie M (2002) Novel KCNJ2 mutation in familial periodic paralysis with ventricular dysrhythmia. Circulation 105:2592–2594. doi:10.1161/01.CIR.0000019906.35135.A3
Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Benson DW (2002) KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet 71:663–668. doi:10.1086/342360
Ballester LY, Benson DW, Wong B, Law IH, Mathews KD, Vanoye CG, George AL (2006) Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum Mutat 27:388. doi:10.1002/humu.9418
Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, Wilde AA, McNitt S, Peterson DR, Zareba W, Robinson JL, Ackerman MJ, Cypress M, Gray DA, Hofman N, Kanters JK, Kaufman ES, Platonov PG, Qi M, Towbin JA, Vincent GM, Lopes CM (2012) Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation 125:1988–1996. doi:10.1161/CIRCULATIONAHA.111.048041
Bendahhou S, Donaldson MR, Plaster NM, Tristani-Firouzi M, Fu YH, Ptacek LJ (2003) Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J Biol Chem 278:51779–51785. doi:10.1074/jbc.M310278200
Bendahhou S, Fournier E, Gallet S, Menard D, Larroque MM, Barhanin J (2007) Corticosteroid-exacerbated symptoms in an Andersen’s syndrome kindred. Hum Mol Genet 16:900–906. doi:10.1093/hmg/ddm034
Dassau L, Conti LR, Radeke CM, Ptacek LJ, Vandenberg CA (2011) Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem 286:9526–9541. doi:10.1074/jbc.M110.170597
Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG (2005) Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology 65:1083–1089. doi:10.1212/01.wnl.0000178888.03767.74
Decher N, Renigunta V, Zuzarte M, Soom M, Heinemann SH, Timothy KW, Keating MT, Daut J, Sanguinetti MC, Splawski I (2007) Impaired interaction between the slide helix and the C-terminus of Kir2.1: a novel mechanism of Andersen syndrome. Cardiovasc Res 75:748–757. doi:10.1016/j.cardiores.2007.05.010
Doi T, Makiyama T, Morimoto T, Haruna Y, Tsuji K, Ohno S, Akao M, Takahashi Y, Kimura T, Horie M (2011) A novel KCNJ2 nonsense mutation, S369X, impedes trafficking and causes a limited form of Andersen-Tawil syndrome. Circ Cardiovasc Genet 4:253–260. doi:10.1161/CIRCGENETICS.110.958157
Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu Y-H, Ptácek LJ (2003) PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 60:1811–1816. doi:10.1212/01.WNL.0000072261.14060.47
Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, Chivoret G, Schwartz K, Coumel P, Guicheney P (1997) KvLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation 96:2778–2781. doi:10.1161/01.CIR.96.9.2778
Donner BC, Schullenberg M, Geduldig N, Huning A, Mersmann J, Zacharowski K, Kovacevic A, Decking U, Aller MI, Schmidt KG (2011) Functional role of TASK-1 in the heart: studies in TASK-1-deficient mice show prolonged cardiac repolarization and reduced heart rate variability. Basic Res Cardiol 106:75–87. doi:10.1007/s00395-010-0128-x
Eckhardt LL, Farley AL, Rodriguez E, Ruwaldt K, Hammill D, Tester DJ, Ackerman MJ, Makielski JC (2007) KCNJ2 mutations in arrhythmia patients referred for LQT testing: a mutation T305A with novel effect on rectification properties. Heart Rhythm 4:323–329. doi:10.1016/j.hrthm.2006.10.025
Fink M, Duprat F, Heurteaux C, Lesage F, Romey G, Barhanin J, Lazdunski M (1996) Dominant negative chimeras provide evidence for homo and heteromultimeric assembly of inward rectifier K+ channel proteins via their N-terminal end. FEBS Lett 378:64–68. doi:10.1016/0014-5793(95)01388-1
Haruna Y, Kobori A, Makiyama T, Yoshida H, Akao M, Doi T, Tsuji K, Ono S, Nishio Y, Shimizu W, Inoue T, Murakami T, Tsuboi N, Yamanouchi H, Ushinohama H, Nakamura Y, Yoshinaga M, Horigome H, Aizawa Y, Kita T, Horie M (2007) Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum Mutat 28:208. doi:10.1002/humu.9483
Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, Nishio Y, Sasaki K, Itoh H, Yokode M, Kita T, Horie M, Kimura T (2012) A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res 93:666–673. doi:10.1093/cvr/cvr329
Hosaka Y, Hanawa H, Washizuka T, Chinushi M, Yamashita F, Yoshida T, Komura S, Watanabe H, Aizawa Y (2003) Function, subcellular localization and assembly of a novel mutation of KCNJ2 in Andersen’s syndrome. J Mol Cell Cardiol 35:409–415. doi:10.1016/S0022-2828(03)00046-4
Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, Wu J, Ohno S, Makiyama T, Miyamoto A, Naiki N, Wang Q, **e Y, Suzuki T, Tateno S, Nakamura Y, Zang WJ, Ito M, Matsuura H, Horie M (2012) Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet 5:344–353. doi:10.1161/CIRCGENETICS.111.962316
Liu Y, Sun L, Pan Z, Bai Y, Wang N, Zhao J, Xu C, Li Z, Li B, Du Z, Lu Y, Gao X, Yang B (2011) Overexpression of M3 muscarinic receptor is a novel strategy for preventing sudden cardiac death in transgenic mice. Mol Med 17:1179–1187. doi:10.2119/molmed.2011.00093
Lopatin AN, Makhina EN, Nichols CG (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372:366–369. doi:10.1038/372366a0
Lopes CMB, Zhang H, Rohacs T, ** T, Yang J, Logothetis DE (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34:933–944. doi:10.1016/S0896-6273(02)00725-0
Matsuda H, Saigusa A, Irisawa H (1987) Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature 325:156–159. doi:10.1038/325156a0
Modoni A, Bianchi ML, Vitulano N, Pagliarani S, Perna F, Sanna T, Rizzo V, Silvestri G (2011) Lack of any cardiac involvement in a patient with Andersen-Tawil syndrome associated with the c.574A→G mutation in KCNJ2. Cardiology 120:200–203. doi:10.1159/000335529
Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8:279–287. doi:10.1038/nn1411
Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R (2001) Mutations in Kir2.1 Cause the Developmental and Episodic Electrical Phenotypes of Andersen’s Syndrome. Cell 105:511–519. doi:10.1016/S0092-8674(01)00342-7
Preisig-Müller R, Schlichthörl G, Goerge T, Heinen S, Brüggemann A, Rajan S, Derst C, Veh RW, Daut J (2002) Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome. Proc Natl Acad Sci USA 99:7774–7779. doi:10.1073/pnas.102609499
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96:800–807. doi:10.1161/01.RES.0000162101.76263.8c
Raab-Graham KF, Radeke CM, Vandenberg CA (1994) Molecular cloning and expression of a human heart inward rectifier potassium channel. NeuroReport 5:2501–2505. doi:10.1097/00001756-199412000-00024
Ryan DP, da Silva MR, Soong TW, Fontaine B, Donaldson MR, Kung AW, Jongjaroenprasert W, Liang MC, Khoo DH, Cheah JS, Ho SC, Bernstein HS, Maciel RM, Brown RH Jr, Ptacek LJ (2010) Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140:88–98. doi:10.1016/j.cell.2009.12.024
Schram G, Melnyk P, Pourrier M, Wang Z, Nattel S (2002) Kir2.4 and Kir2.1 K+ channel subunits co-assemble: a potential new contributor to inward rectifier current heterogeneity. J Physiol 544:337–349. doi:10.1113/jphysiol.2002.026047
Seebohm G, Strutz-Seebohm N, Ursu ON, Preisig-Müller R, Zuzarte M, Hill EV, Kienitz MC, Bendahhou S, Fauler M, Tapken D, Decher N, Collins A, Jurkat-Rott K, Steinmeyer K, Lehmann-Horn F, Daut J, Tavare JM, Pott L, Bloch W, Lang F (2012) Altered stress stimulation of inward rectifier potassium channels in Andersen-Tawil syndrome. FASEB J 26:513–522. doi:10.1096/fj.11-189126
Tinker A, Jan YN, Jan LY (1996) Regions responsible for the assembly of inwardly rectifying potassium channels. Cell 87:857–868. doi:10.1016/S0092-8674(00)81993-5
Tristani-Firouzi M, Etheridge SP (2010) Kir2.1 channelopathies: the Andersen-Tawil syndrome. Pflügers Arch 460:289–294. doi:10.1007/s00424-010-0820-6
Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu Y-H, Ptacek LJ, Tawil R (2002) Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 110:381–388. doi:10.1172/JCI15183
World medical association, American physiological society (2002) Guiding principles for research involving animals and human beings. Am J Physiol Regul Integr Comp Physiol 283:R281–283. doi:10.1152/ajpregu.00279.2002
Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE (2011) Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471:230–234. doi:10.1038/nature09855
Zuzarte M, Rinné S, Schlichthörl G, Schubert A, Daut J, Preisig-Müller R (2007) A di-acidic sequence motif enhances the surface expression of the potassium channel TASK-3. Traffic 8:1093–1100. doi:10.1111/j.1600-0854.2007.00593.x
Acknowledgments
We would like to thank Kirsten Ramlow and Oxana Nowak for excellent technical support, Ruth Bennett, the ExoSeq team, the staff of the WTSI for their invaluable contribution to the manuscript. This work was supported by Deutsche Forschungs-gemeinschaft (DFG) Grant DE1482-3/2 to N.D., The Fondation Leducq, Paris (to E. S.-B.), and by the Wellcome Trust (Grant number 098051).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. M. Limberg and S. Zumhagen contributed equally.
Niels Decher and Eric Schulze-Bahr are co-senior authors.
Rights and permissions
About this article
Cite this article
Limberg, M.M., Zumhagen, S., Netter, M.F. et al. Non dominant-negative KCNJ2 gene mutations leading to Andersen-Tawil syndrome with an isolated cardiac phenotype. Basic Res Cardiol 108, 353 (2013). https://doi.org/10.1007/s00395-013-0353-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-013-0353-1