
Abstract
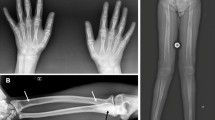
Disorders of post-squalene cholesterol biosynthesis are inborn errors of metabolism characterised by multiple congenital abnormalities, including significant skeletal involvement. The most frequent and best-characterised example is the Smith–Lemli–Opitz syndrome. Nine other disorders are known, namely autosomal-recessive Antley–Bixler syndrome, Greenberg dysplasia, X-linked dominant chondrodysplasia punctata, X-linked recessive male emopamil-binding protein deficiency, CHILD syndrome, CK syndrome, sterol C4 methyloxidase-like deficiency, desmosterolosis and lathosterolosis. This study provides an overview of the radiologic features observed in these diseases. A common pattern of limb abnormalities is recognisable, including polydactyly, which is typically post-axial and rarely interdigital and can involve all four limbs, and syndactyly of the toes. Chondrodysplasia punctata is specifically associated with a subgroup of disorders of cholesterol biosynthesis (Greenberg dysplasia, CHILD syndrome, X-linked dominant chondrodysplasia punctata, male emopamil-binding protein deficiency). The possible occurrence of epiphyseal stippling in the Smith–Lemli–Opitz syndrome, initially reported, does not appear to be confirmed. Stippling is also associated with other congenital disorders such as chromosomal abnormalities, brachytelephalangic chondrodysplasia punctata (X-linked recessive chondrodysplasia punctata, disruptions of vitamin K metabolism, maternal autoimmune diseases), rhizomelic chondrodysplasia punctata (peroxisomal disorders) and lysosomal storage disorders. In the differential diagnosis of epiphyseal stippling, a moth-eaten appearance of bones, asymmetry, or presence of a common pattern of limb abnormalities indicate inborn errors of cholesterol biosynthesis. We highlight the specific differentiating radiologic features of disorders of post-squalene cholesterol biosynthesis.










Similar content being viewed by others


References
Porter FD, Herman GE (2011) Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 52:6–34
Offiah AC, Mansour S, Jeffrey I et al (2003) Greenberg dysplasia (HEM) and lethal X linked dominant Conradi–Hunermann chondrodysplasia punctata (CDPX2): presentation of two cases with overlap** phenotype. J Med Genet 40:e129
Horn LC, Faber R, Meiner A et al (2000) Greenberg dysplasia: first reported case with additional non-skeletal malformations and without consanguinity. Prenat Diagn 20:1008–1011
Konstantinidou A, Karadimas C, Waterham HR et al (2008) Pathologic, radiographic and molecular findings in three fetuses diagnosed with HEM/Greenberg skeletal dysplasia. Prenat Diagn 28:309–312
Trajkovski Z, Vrcakovski M, Saveski J et al (2002) Greenberg dysplasia (hydrops-ectopic calcification-moth-eaten skeletal dysplasia): prenatal ultrasound diagnosis and review of literature. Am J Med Genet 111:415–419
Wassif CA, Brownson KE, Sterner AL et al (2007) HEM dysplasia and ichthyosis are likely laminopathies and not due to 3beta-hydroxysterol Delta14-reductase deficiency. Hum Molec Genet 16:1176–1187
Waterham HR, Koster J, Mooyer P et al (2003) Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet 72:1013–1017
Hoffmann K, Dreger CK, Olins AL et al (2002) Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nat Genet 31:410–414
Oosterwijk JC, Mansour S, van Noort G et al (2003) Congenital abnormalities reported in Pelger-Huet homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J Med Genet 40:937–941
Borovik L, Modaff P, Waterham HR et al (2013) Pelger-huet anomaly and a mild skeletal phenotype secondary to mutations in LBR. Am J Med Genet A 161:2066–2073
Clayton P, Fischer B, Mann A et al (2010) Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus 1:354–366
Chitayat D, Gruber H, Mullen BJ et al (1993) Hydrops-ectopic calcification-moth-eaten skeletal dysplasia (Greenberg dysplasia): prenatal diagnosis and further delineation of a rare genetic disorder. Am J Med Genet 47:272–277
Bouvier R, Cordier M, Loget P et al (2008) Maladies métaboliques. In: Pathologie fœtale et placentaire pratique. Sauramps Médical, Montpellier, France, pp 465–484
Hall CM, Offiah A, Forzano F et al (2012) Fetal and perinatal skeletal dysplasias: an atlas of multimodality imaging. Radcliffe Publishing Ltd., London, UK
Derry JM, Gormally E, Means GD et al (1999) Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet 22:286–290
Braverman N, Lin P, Moebius FF et al (1999) Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet 22:291–294
Tysoe C, Law CJ, Caswell R et al (2008) Prenatal testing for a novel EBP missense mutation causing X-linked dominant chondrodysplasia punctata. Prenat Diagn 28:384–388
Whittock NV, Izatt L, Simpson-Dent SL et al (2003) Molecular prenatal diagnosis in a case of an X-linked dominant chondrodysplasia punctata. Prenat Diagn 23:701–704
Kelley RI, Wilcox WG, Smith M et al (1999) Abnormal sterol metabolism in patients with Conradi–Hunermann–Happle syndrome and sporadic lethal chondrodysplasia punctata. Am J Med Genet 83:213–219
Akiyama M, Sakai K, Hayasaka K et al (2009) Conradi–Hunermann–Happle syndrome with abnormal lamellar granule contents. Br J Dermatol 160:1335–1337
Umranikar S, Glanc P, Unger S et al (2006) X-linked dominant chondrodysplasia punctata: prenatal diagnosis and autopsy findings. Prenat Diagn 26:1235–1240
DiPreta EA, Smith KJ, Skelton H (2000) Cholesterol metabolism defect associated with Conradi–Hunerman–Happle [stet] syndrome. Int J Dermatol 39:846–850
Steijlen PM, van Geel M, Vreeburg M et al (2007) Novel EBP gene mutations in Conradi–Hunermann–Happle syndrome. Br J Dermatol 157:1225–1229
Herman GE, Kelley RI, Pureza V et al (2002) Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome). Genet Med 4:434–438
Has C, Bruckner-Tuderman L, Muller D et al (2000) The Conradi–Hunermann–Happle syndrome (CDPX2) and emopamil binding protein: novel mutations, and somatic and gonadal mosaicism. Hum Mol Genet 9:1951–1955
Rakheja D, Read CP, Hull D et al (2007) A severely affected female infant with X-linked dominant chondrodysplasia punctata: a case report and a brief review of the literature. Pediatr Dev Pathol 10:142–148
Whittock NV, Izatt L, Mann A et al (2003) Novel mutations in X-linked dominant chondrodysplasia punctata (CDPX2). J Invest Dermatol 121:939–942
Hoang MP, Carder KR, Pandya AG et al (2004) Ichthyosis and keratotic follicular plugs containing dystrophic calcification in newborns: distinctive histopathologic features of X-linked dominant chondrodysplasia punctata (Conradi–Hunermann–Happle syndrome). Am J Dermatopathol 26:53–58
Shirahama S, Miyahara A, Kitoh H et al (2003) Skewed X-chromosome inactivation causes intra-familial phenotypic variation of an EBP mutation in a family with X-linked dominant chondrodysplasia punctata. Hum Genet 112:78–83
Grange DK, Kratz LE, Braverman NE et al (2000) CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet 90:328–335
Happle R, Konig A, Grzeschik KH (2000) Behold the CHILD, it’s only one: CHILD syndrome is not caused by deficiency of 3 beta-hydroxysteroid-Delta 8, Delta 7-isomerase. Am J Med Genet 94:341–343
Ausavarat S, Tanpaiboon P, Tongkobpetch S et al (2008) Two novel EBP mutations in Conradi–Hunermann–Happle syndrome. Eur J Dermatol 18:391–393
Hellenbroich Y, Grzeschik KH, Krapp M et al (2007) Reduced penetrance in a family with X-linked dominant chondrodysplasia punctata. Eur J Med Gen 50:392–398
Ikegawa S, Ohashi H, Ogata T et al (2000) Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet 94:300–305
Aughton DJ, Kelley RI, Metzenberg A et al (2003) X-linked dominant chondrodysplasia punctata (CDPX2) caused by single gene mosaicism in a male. Am J Med Genet A 116A:255–260
Arnold AW, Bruckner-Tuderman L, Has C et al (2012) Conradi–Hunermann–Happle syndrome in males vs. MEND syndrome (male EBP disorder with neurological defects). Br J Dermatol 166:1309–1313
Sutphen R, Amar MJ, Kousseff BG et al (1995) XXY male with X-linked dominant chondrodysplasia punctata (Happle syndrome). Am J Med Genet 57:489–492
Furtado LV, Bayrak-Toydemir P, Hulinsky B et al (2010) A novel X-linked multiple congenital anomaly syndrome associated with an EBP mutation. Am J Med Genet A 152A:2838–2844
Milunsky JM, Maher TA, Metzenberg AB (2003) Molecular, biochemical, and phenotypic analysis of a hemizygous male with a severe atypical phenotype for X-linked dominant Conradi–Hunermann–Happle syndrome and a mutation in EBP. Am J Med Genet A 116:249–254
Herman GE, Kratz L (2012) Disorders of sterol synthesis: beyond Smith–Lemli–Opitz syndrome. Am J Med Genet C: Semin Med Genet 160C:301–321
Happle R (2011) A novel X-linked phenotype caused by hypomorphic EBP mutations. Am J Med Genet A 155:1770–1771
Kelley RI, Kratz LE, Glaser RL et al (2002) Abnormal sterol metabolism in a patient with Antley–Bixler syndrome and ambiguous genitalia. Am J Med Genet 110:95–102
Fluck CE, Tajima T, Pandey AV et al (2004) Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley–Bixler syndrome. Nat Genet 36:228–230
Konig A, Happle R, Bornholdt D et al (2000) Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet 90:339–346
Bornholdt D, Konig A, Happle R et al (2005) Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet 42:e17
Ishibashi M, Matsuda F, Oka H et al (2006) Abnormal lamellar granules in a case of CHILD syndrome. J Cutan Pathol 33:447–453
Happle R (2010) The group of epidermal nevus syndromes part I. Well defined phenotypes. J Am Acad Dermatol 63:1–22
McLarren KW, Severson TM, du Souich C et al (2010) Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am J Hum Genet 87:905–914
Du Souich C, Chou A, Yin J et al (2009) Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus. Am J Med Genet A 149:2469–2478
Tarpey PS, Smith R, Pleasance E et al (2009) A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 41:535–543
He M, Kratz LE, Michel JJ et al (2011) Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J Clin Invest 121:976–984
Rossi M, D’Armiento M, Parisi I et al (2007) Clinical phenotype of lathosterolosis. Am J Med Genet A 143:2371–2381
Kelley RI, Hennekam RC (2000) The Smith–Lemli–Opitz syndrome. J Med Genet 37:321–335
Ryan AK, Bartlett K, Clayton P et al (1998) Smith–Lemli–Opitz syndrome: a variable clinical and biochemical phenotype. J Med Genet 35:558–565
Quelin C, Loget P, Verloes A et al (2012) Phenotypic spectrum of fetal Smith–Lemli–Opitz syndrome. Eur J Med Genet 55:81–90
De Jong G, Kirby PA, Muller LM (1998) RSH (Smith–Lemli–Opitz) syndrome: ‘severe’ phenotype with ectrodactyly. Am J Med Genet 75:283–287
Warman ML, Cormier-Daire V, Hall C et al (2011) Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 155:943–968
Wallace M, Zori RT, Alley T et al (1994) Smith–Lemli–Opitz syndrome in a female with a de novo, balanced translocation involving 7q32: probable disruption of an SLOS gene. Am J Med Genet 50:368–374
Nowaczyk MJ (2011) Epiphyseal stippling is not a feature of 7-dehydrocholesterol reductase deficiency (Smith–Lemli–Opitz syndrome). Am J Med Genet A 155:940–941
Brunetti-Pierri N, Corso G, Rossi M et al (2002) Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3beta-hydroxysteroid-delta5-desaturase. Am J Hum Genet 71:952–958
Rossi M, Vajro P, Iorio R et al (2005) Characterization of liver involvement in defects of cholesterol biosynthesis: long-term follow-up and review. Am J Med Genet A 132:144–151
Parnes S, Hunter AG, Jimenez C et al (1990) Apparent Smith–Lemli–Opitz syndrome in a child with a previously undescribed form of mucolipidosis not involving the neurons. Am J Med Genet 35:397–405
Krakowiak PA, Wassif CA, Kratz L et al (2003) Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum Mol Genet 12:1631–1641
Ho AC, Fung CW, Siu TS et al (2014) Lathosterolosis: a disorder of cholesterol biosynthesis resembling Smith–Lemli–Opitz syndrome. JIMD Rep 12:129–134
Schaaf CP, Koster J, Katsonis P et al (2011) Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet A 155:1597–1604
Andersson HC, Kratz L, Kelley R (2002) Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet 113:315–319
Waterham HR, Koster J, Romeijn GJ et al (2001) Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet 69:685–694
FitzPatrick DR, Keeling JW, Evans MJ et al (1998) Clinical phenotype of desmosterolosis. Am J Med Genet 75:145–152
Clayton P, Mills K, Keeling J et al (1996) Desmosterolosis: a new inborn error of cholesterol biosynthesis. Lancet 348:404
Villavicencio EH, Walterhouse DO, Iannaccone PM (2000) The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet 67:1047–1054
Irving MD, Chitty LS, Mansour S et al (2008) Chondrodysplasia punctata: a clinical diagnostic and radiological review. Clin Dysmorphol 17:229–241
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rossi, M., Hall, C.M., Bouvier, R. et al. Radiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis. Pediatr Radiol 45, 965–976 (2015). https://doi.org/10.1007/s00247-014-3257-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-014-3257-9