
Abstract
Background
Lipid pathways play a crucial role in psoriatic arthritis development, and some lipid-lowering drugs are believed to have therapeutic benefits due to their anti-inflammatory properties. Traditional observational studies face issues with confounding factors, complicating the interpretation of causality. This study seeks to determine the genetic link between these medications and the risk of psoriatic arthritis.
Methods
This drug target study utilized the Mendelian randomization strategy. We harnessed high-quality data from population-level genome-wide association studies sourced from the UK Biobank and FinnGen databases. The inverse variance-weighted method, complemented by robust pleiotropy methods, was employed. We examined the causal relationships between three lipid-lowering agents and psoriatic arthritis to unveil the underlying mechanisms.
Results
A significant association was observed between genetically represented proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition and a decreased risk of psoriatic arthritis (odds ratio [OR]: 0.51; 95% CI 0.14–0.88; P < 0.01). This association was further corroborated in an independent dataset (OR 0.60; 95% CI 0.25–0.94; P = 0.03). Sensitivity analyses affirmed the absence of statistical evidence for pleiotropic or genetic confounding biases. However, no substantial associations were identified for either 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors or Niemann–Pick C1-like 1 inhibitors.
Conclusions
This Mendelian randomization analysis underscores the pivotal role of PCSK9 in the etiology of psoriatic arthritis. Inhibition of PCSK9 is associated with reduced psoriatic arthritis risk, highlighting the potential therapeutic benefits of existing PCSK9 inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory disorder that primarily targets joints and various components of the musculoskeletal system [1]. Recent studies indicate that psoriatic arthritis, affecting up to 30% of individuals with psoriasis, has a prevalence of 6–25 cases per 10,000 people in the USA [2]. The clinical presentations of PsA are diverse, encompassing peripheral joint inflammation, inflammatory back discomfort, enthesitis, and tenosynovitis [3]. The debilitating effects of PsA are now recognized to be on par with other inflammatory arthritic conditions such as rheumatoid arthritis (RA) and axial spondyloarthritides (axSpA) [2]. The functional impairments stemming from PsA often result in decreased work efficiency, increased absenteeism, and a marked decline in the quality of life for those affected [4].
A significant observation among PsA patients is the prevalence of disrupted lipid metabolism, which elevates the risk of cardiovascular complications [5,6,7]. Thus, screening for dyslipidemia becomes imperative for those diagnosed with PsA [8, 9]. While lipid-lowering medications are pivotal in managing cardiovascular risks [10], their direct impact on PsA treatment remains a topic of ongoing research. The efficacy of several mainstream lipid-lowering drugs in treating PsA symptoms is yet to be conclusively established.
Exploring lipid pathway interventions in PsA is compelling due to several factors. Some lipid-lowering medications exhibit anti-inflammatory properties, suggesting their potential as therapeutic agents for PsA. The unclear pathogenesis of PsA highlights the importance of identifying lipid-related causal pathways. Using lipid-lowering drugs that target both lipid imbalances and PsA offers personalized treatment options, especially for those with a strong family history and dyslipidemia. If these drugs show disease-modifying effects, they could be repurposed for PsA treatment, reducing immunosuppression risks. However, traditional pharmacoepidemiologic designs present challenges in obtaining solid evidence.
The inherent genetic variations related to protein drug targets can shed light on potential clinical outcomes [11]. Leveraging genetic instrumental variable analyses, or Mendelian randomization (MR), offers a quasi-randomized approach, providing a more resilient framework against biases typically seen in conventional epidemiological studies [12]. Our study's objective was to employ a two-sample MR to delve into the relationship between PsA risk and three genetically indicated lipid-lowering drugs: proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (eg, alirocumab), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) inhibitors (ie, statins), and Niemann–Pick C1-like 1 (NPC1L1) inhibitors (i.e., ezetimibe).
Materials and methods
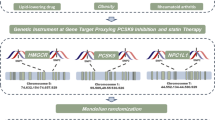
This research employed deidentified summary data derived from prior genome-wide association studies (GWAS). Ethical clearances were secured for all original investigations, and relevant references are comprehensively documented. The study design is illustrated in Fig. 1.

Research overview and design of drug target Mendelian randomization analysis. To establish a causal link, the following criteria must be met: (1) instrumental variables should be independent of confounders (indicated by dashed lines), (2) instrumental variables must be associated with the exposure (depicted by solid lines), and (3) instrumental variables shouldn't have a direct connection to the outcome (represented by dashed lines). PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; NPC1L1, Niemann-Pick C1-like 1; LDL, low-density lipoprotein
Genetic proxies for lipid-lowering drugs
We selected low-density lipoprotein (LDL) as a biomarker due to the demonstrated efficacy of three lipid-lowering drugs in reducing LDL cholesterol levels. To the best of our knowledge, we utilized the most comprehensive GWAS meta-analysis currently available, which has established genetic associations with LDL, covering 12,321,875 SNPs with a sample size of 440,546 [13].
We identified variants associated with LDL at a genome-wide significance level (P < 5 × 10−8). These variants exhibited minimal correlation, with a linkage disequilibrium threshold of r2 < 0.3, as determined using PLINK and referencing phase 3 version 5 of the 1000 Genomes Project. We focused on regions within ± 100 kb of the PCSK9 gene (build GRCh37/hg19: chromosome 1: 55,505,221, 55,530,525) for PCSK9 inhibitors (e.g., alirocumab or evolocumab), the HMGCR gene (chromosome 5: 74,632,154, 74,657,929) as an instrumental variable for statins, and the NPC1L1 gene (chromosome 7: 44,552,134, 44,580,914) as an instrumental variable for ezetimibe.
Considering the observed correlation between lipids and PsA, we explored the potential association between genetically predicted LDL levels and the risk of PsA. To instrument LDL, we utilized genome-wide significant variants with pairwise correlations below r2 < 0.001, excluding variants from the three previously mentioned drug target gene regions.
Genetic association for psoriatic arthritis
We obtained genetic associations for PsA from the GWAS data with the largest number of SNPs in the FinnGen database (eTable 1). This dataset has a sample size of 213,879, which includes 16,380,462 SNPs, 1637 patients, and 212,242 controls. PsA is defined according to the standards in the International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10). The ICD-10 code is L40.5.
To validate our analyses, we sourced genetic associations for PsA from an alternative dataset comprising a sample size of 218,792, encompassing 1455 patients and 217,337 controls (eTable 1).
Statistical analysis and MR assumptions
We employed the inverse variance-weighted approach with multiplicative random effects to derive a weighted mean from individual variant estimates [14]. A valid instrumental variable adheres to three fundamental assumptions [15]. Firstly, the variants should exhibit a significant association with the intended exposure. We calculated the F statistics for drug target instruments, using the ratio of the squared β to the squared standard error. An F statistic exceeding 10 indicates a sufficiently robust instrument strength [16].
Secondly, there should be no mutual causal relationship between the variants and the outcome. To mitigate confounding due to inherent population structures, we restricted our analysis to populations of European descent.
Thirdly, the variants should influence the outcome solely through the specified risk factor. To assess the resilience of our primary inverse variance-weighted estimates against horizontal pleiotropy (where instruments influence the outcome via factors other than the exposure), we employed the MR Egger [17], weighted median [18], and weighted mode [19] methodologies. We referenced the PhenoScanner [20, 21], a meticulously curated genotype–phenotype database, to identify correlations between variants designated for each drug target and other potential traits indicative of pleiotropic pathways. Notably, traits linked to smoking [22] and psoriasis [23], which are established risk determinants for PsA disease, were scrutinized. In our sensitivity analyses, variants that exhibited associations with these and other relevant traits, surpassing the threshold of P < 1 × 10−5, were excluded to mitigate potential sources of pleiotropy.
Supplementary analysis
To ensure the robustness of our primary analyses, we conducted a supplementary evaluation. We validated our instruments by employing coronary artery disease as a positive control outcome, considering the well-established therapeutic effects of lipid-lowering drugs in this domain. Genetic associations were sourced from a GWAS encompassing 42,096 clinically validated cases (e.g., myocardial infarction, acute coronary syndrome, chronic stable angina, or coronary stenosis exceeding 50%) [24].We also used data from another study for validation [25].
Results
In primary analysis utilizing the PsA dataset from the FinnGen database, we identified 33 genetic variants representing LDL reduction via PCSK9 inhibition (with a mean F statistic of 167), 19 variants for HMGCR (with a mean F statistic of 135), and 6 for NPC1L1 (with a mean F statistic of 72) as detailed in Table 1.
Exp, exposure; Chr, chromosome; Pos, position; EA, effect allele; OA, other allele; EAF, effect allele frequency; Se, standard error; PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A; NPC1L1, Niemann–Pick C1-Like 1; SNP, single-nucleotide polymorphism
Our findings indicate that genetically proxied PCSK9 inhibition correlates with a decreased PsA risk (odds ratio [OR]: 0.51; 95% CI 0.14–0.88; P < 0.01). This association was validated using an alternative GWAS dataset (OR 0.60; 95% CI 0.25–0.94; P = 0.03). There was no statistical heterogeneity in both the primary (P = 0.97) and validation estimates (P = 0.93), as depicted in Fig. 2, 3. Our sensitivity analyses yielded congruent results, with no discernible bias from horizontal pleiotropy, as illustrated in Fig. 3, 4 and eTable 2, 3.

Associations between genetically proxied lipid-lowering drugs and psoriatic arthritis risk. PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; NPC1L1, Niemann–Pick C1-like 1; LDL, low-density lipoprotein; OR, odds ratio; PsA, psoriatic arthritis

Summary of psoriatic arthritis risk results from pleiotropy robust sensitivity analyses. PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; NPC1L1, Niemann–Pick C1-like 1; LDL, low-density lipoprotein; IVW, inverse variance-weighted method; OR, odds ratio

Sensitivity analysis of lipid-lowering drugs and LDL on psoriatic arthritis. Leave-one-out analysis of (A) PCSK9 on PsA, (B) PCSK9 on PsA (validation), (C) HMGCR on PsA, (D) HMGCR on PsA (validation), (E) NPC1L1 on PsA, (F) NPC1L1 on PsA (validation), (G) LDL on PsA, (H) LDL on PsA (validation). PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; NPC1L1, Niemann–Pick C1-like 1; LDL, low-density lipoprotein; PsA, psoriatic arthritis
Conversely, there was minimal evidence linking HMGCR with PsA risk across datasets (Figs. 2 and 3). The results for NPC1L1 mirrored this observation. Furthermore, the genetically inferred reduction in LDL, instrumented by 177 genetic variants (mean F statistic of 166) (eTable 4), showed no association with PsA in both primary and validation analyses.
The genetic proxy for the inhibition of all three drug targets, as well as LDL, consistently demonstrated an association with a decreased risk of the positive control outcome, coronary artery disease, as illustrated in Fig. 5 and eTable 5. This association was evident in both the primary and validation analyses.

Correlations between genetically inferred lipid-lowering medications and coronary artery disease risk—positive control analysis. PCSK9, proprotein convertase subtilisin/kexin type 9; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; NPC1L1, Niemann-Pick C1-like 1; LDL, low-density lipoprotein; CAD, coronary artery disease; OR, odds ratio
Discussion
In this large-scale MR analysis, we delved into the impacts of three prevalent LDL-reducing drug targets (PCSK9 inhibitors, HMGCR—the target of statins, and NPC1L1 inhibitors—the target of ezetimibe) on the risks of PsA. Our findings underscore a causal link between PCSK9 inhibition and a diminished PsA risk, a relationship that seems independent of circulating LDL concentrations, as no overarching LDL–PsA risk association was discerned. Notably, genetic proxies for HMGCR and NPC1L1 inhibition showed no causal relationship with PsA risk.
PCSK9, a post-translational modulator of the LDL receptor (LDLR), orchestrates LDLR internalization and subsequent degradation. Its significance as a pivotal therapeutic target for hypercholesterolemia and coronary heart disease attenuation is evident [26]. A clinical study showed that the levels of PCSK9 in serum were moderately to severely correlated with the levels of LDL and total cholesterol (TChol) [27]. The FOURIER study demonstrated that PCSK9 inhibitors profoundly expunge LDL cholesterol from the circulatory system, leading to attenuated cardiovascular adversities [28].
While PCSK9's lipid-regulating function is universally acknowledged, its multifaceted functions extend to inflammation, gastrointestinal diseases, and viral infections [29,30,31]. Its role in inflammation, albeit pivotal, has often been relegated to the background. Research has pinpointed heightened PCSK9 expression elevating inflammatory chemokine production, including interleukin-1α, interleukin-6, and tumor necrosis factor-α (TNF-α) [32]. PCSK9 modulates the expression of TNF receptor-associated factors via the nuclear factor kappa-light-chain enhancer of activated B cells(NF-κB) signaling pathway [33, 34]. Moreover, PCSK9 is intertwined with several inflammatory pathways, encompassing the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, TNF-α, and resistin [35, 36].
PCSK9 not only plays a crucial role in the regulation of serum LDL levels, but its biological function seems to extend beyond the regulation of cholesterol metabolism [37]. Current research indicates that PCSK9 is a key regulatory factor in the inflammation of chronic and autoimmune diseases [38]. In certain chronic conditions, such as chronic kidney disease and hypothyroidism, the expression and release of PCSK9 are also increased during the inflammatory process [39, 40].
Although the exact etiology of PsA remains elusive, prevailing research underscores the pivotal role of immune and inflammatory factors in its pathogenesis [3, 41]. PCSK9-mediated inflammatory cascades might be instrumental in the pathophysiological orchestration of PsA. Elevated serum PCSK9 concentrations have been documented in PsA cohorts [42]. PCSK9's potential to galvanize macrophage activation is noteworthy [43, 44]. Such macrophage activation, a hallmark of PsA pathogenesis, instigates a cascade of cytokines, notably Interleukin-17(IL-17), IL-1, TNF-α, and IL-23, which incite joint and entheseal inflammation, leading to cartilaginous and osseous degradation [45,46,47]. IL-17, pivotal in the inflammatory cascade, tissue damage, and bone erosion linked to PsA, has been spotlighted [48]. Elevated IL-17 levels and type 3 innate lymphoid cells, IL-17 producers, have been identified in PsA patient synovial fluid [49]. In hyperlipidemic mouse models, PCSK9 knockout reduced circulating IL-17 levels and the differentiation of IL-17-producing cells [50]. TNFα and IL1-β, pivotal inflammatory mediators in PsA, are suppressed in PCSK9-inhibited macrophages post-lipid exposure [51]. In conclusion, the data presented elucidates a significant association between PCSK9 and the pathophysiology of PsA. Coupled with our study outcomes, PCSK9 emerges as a promising therapeutic target.
Statins, ubiquitously employed lipid-lowering agents, are also prevalent among PsA patients to mitigate cardiovascular risk [52, 53]. Previous observational studies have probed the ramifications of statins on PsA's clinical trajectory, albeit with inconsistent outcomes [54,55,56]. Our study offers a resolution to this conundrum, revealing no causal ties between HMCGR inhibition and PsA susceptibility.
NPC1L1, a cholesterol uptake transporter protein predominantly expressed in the small intestine and liver. NPC1L1 inhibitor is another widely cited lipid-lowering agent [57]. To our knowledge, no extant research has elucidated NPC1L1 inhibitors causal relationship with PsA. Our findings indicate that genetic proxies for NPC1L1 inhibition remain unassociated with PsA, underscoring the need for further clinical validation.
This study, to the best of our knowledge, pioneers the MR approach to discern the causal effects of lipid-lowering agents on PsA. The robustness of our results, validated through replication and sensitivity analyses, is a notable strength. Nonetheless, this study has some limitations. Primarily, the MR analysis is susceptible to biases stemming from potential deviations from standard instrumental variable assumptions. Yet, multiple sensitivity analyses within our study found no indications of such violations, reinforcing the integrity of our primary results. Additionally, our study cohort was exclusively of European descent, underscoring the need for subsequent research in diverse ethnic groups to enhance the external validity of our conclusions.
Conclusions
Our study elucidates that genetically proxied PCSK9 inhibition is inversely associated with psoriatic arthritis susceptibility, underscoring the therapeutic potential of extant PCSK9 inhibitors. Furthermore, this research suggests the possibility of personalized lipid-lowering drug selections for those with a predisposition to psoriatic arthritis. To cement these findings, subsequent randomized controlled trials are imperative.
Data availability
The GWAS summary statistics data used in this MR study is available in OpenGWAS (https://gwas.mrcieu.ac.uk/).
References
Lee B-W, Moon S-J. Inflammatory Cytokines in Psoriatic Arthritis: Understanding Pathogenesis and Implications for Treatment. Int J Mol Sci. 2023;24:11662.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic Arthritis. N Engl J Med. 2017;376:957–70.
Veale DJ, Fearon U. The pathogenesis of psoriatic arthritis. Lancet. 2018;391:2273–84.
Haroon M, Gallagher P, FitzGerald O. Diagnostic delay of more than 6 months contributes to poor radiographic and functional outcome in psoriatic arthritis. Ann Rheum Dis. 2015;74:1045–50.
Ma C, Schupp CW, Armstrong EJ, Armstrong AW. Psoriasis and dyslipidemia: a population-based study analyzing the National Health and Nutrition Examination Survey (NHANES). J Eur Acad Dermatol Venereol. 2014;28:1109–12.
Zeng C, Wen B, Hou G, Lei L, Mei Z, Jia X, et al. Lipidomics profiling reveals the role of glycerophospholipid metabolism in psoriasis. Gigascience. 2017;6:1–11.
Pietrzak A, Chabros P, Grywalska E, Kiciński P, Pietrzak-Franciszkiewicz K, Krasowska D, et al. Serum lipid metabolism in psoriasis and psoriatic arthritis - an update. Arch Med Sci. 2019;15:369–75.
Wójcik P, Biernacki M, Wroński A, Łuczaj W, Waeg G, Žarković N, et al. Altered Lipid Metabolism in Blood Mononuclear Cells of Psoriatic Patients Indicates Differential Changes in Psoriasis Vulgaris and Psoriatic Arthritis. Int J Mol Sci. 2019;20:4249.
Taheri Sarvtin M, Hedayati MT, Shokohi T, HajHeydari Z. Serum lipids and lipoproteins in patients with psoriasis. Arch Iran Med. 2014;17:343–6.
Pencina MJ, Pencina KM, Lloyd-Jones D, Catapano AL, Thanassoulis G, Sniderman AD. The Expected 30-Year Benefits of Early Versus Delayed Primary Prevention of Cardiovascular Disease by Lipid Lowering. Circulation. 2020;142:827–37.
Gill D, Georgakis MK, Walker VM, Schmidt AF, Gkatzionis A, Freitag DF, et al. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome Open Res. 2021;6:16.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89-98.
Richardson TG, Sanderson E, Palmer TM, Ala-Korpela M, Ference BA, Davey Smith G, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17: e1003062.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Davies NM, Holmes MV, Davey SG. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362: k601.
Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–64.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016;40:304–14.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–98.
Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32:3207–9.
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–3.
Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369:1379–90.
Stolwijk C, Essers I, van Tubergen A, Boonen A, Bazelier MT, De Bruin ML, et al. The epidemiology of extra-articular manifestations in ankylosing spondylitis: a population-based matched cohort study. Ann Rheum Dis. 2015;74:1373–8.
Nikpay M, Goel A, Won H-H, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30.
Mbatchou J, Barnard L, Backman J, Marcketta A, Kosmicki JA, Ziyatdinov A, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet. 2021;53:1097–103.
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379:2097–107.
Frątczak A, Miziołek B, Łupicka-Słowik A, Sieńczyk M, Polak K, Bergler-Czop B. Significance of Neutrophil Gelatinase-Associated Lipocalin (NGAL) and Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) for the Monitoring of Treatment Response to Cyclosporine in Patients with Psoriasis. Life (Basel). 2023;13:1873.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376:1713–22.
Momtazi-Borojeni AA, Sabouri-Rad S, Gotto AM, Pirro M, Banach M, Awan Z, et al. PCSK9 and inflammation: a review of experimental and clinical evidence. Eur Heart J Cardiovasc Pharmacother. 2019;5:237–45.
Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, et al. Low-Density Lipoprotein Cholesterol Lowering With Evolocumab and Outcomes in Patients With Peripheral Artery Disease: Insights From the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk). Circulation. 2018;137:338–50.
Zhang L, Wang X, Wang M, Sterling NW, Du G, Lewis MM, et al. Circulating Cholesterol Levels May Link to the Factors Influencing Parkinson’s Risk. Front Neurol. 2017;8:501.
Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, et al. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-κB activation in THP-1-derived macrophages. Int J Mol Med. 2012;30:931–8.
Sun HL, Wu YR, Song FF, Gan J, Huang LY, Zhang L, et al. Role of PCSK9 in the Development of Mouse Periodontitis Before and After Treatment: A Double-Edged Sword. J Infect Dis. 2018;217:667–80.
Tang Z-H, Peng J, Ren Z, Yang J, Li T-T, Li T-H, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. 2017;262:113–22.
Cao A, Wu M, Li H, Liu J. Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells. J Lipid Res. 2011;52:518–30.
Cui C-J, Li S, Zhu C-G, Sun J, Du Y, Zhang Y, et al. Enhanced pro-protein convertase subtilisin/kexin type 9 expression by C-reactive protein through p38MAPK-HNF1α pathway in HepG2 cells. J Cell Mol Med. 2016;20:2374–83.
Frątczak A, Polak K, Szczepanek M, Lis-Święty A. The role of proprotein convertase subtilisin/kexin type 9 (PCSK9) in the pathophysiology of psoriasis and systemic lupus erythematosus. Postepy Dermatol Alergol. 2022;39:645–50.
Pirillo A, Bonacina F, Norata GD, Catapano AL. The Interplay of Lipids, Lipoproteins, and Immunity in Atherosclerosis. Curr Atheroscler Rep. 2018;20:12.
Haas ME, Levenson AE, Sun X, Liao W-H, Rutkowski JM, de Ferranti SD, et al. The Role of Proprotein Convertase Subtilisin/Kexin Type 9 in Nephrotic Syndrome-Associated Hypercholesterolemia. Circulation. 2016;134:61–72.
Girona J, Ibarretxe D, Plana N, Guaita-Esteruelas S, Amigo N, Heras M, et al. Circulating PCSK9 levels and CETP plasma activity are independently associated in patients with metabolic diseases. Cardiovasc Diabetol. 2016;15:107.
Schett G, Rahman P, Ritchlin C, McInnes IB, Elewaut D, Scher JU. Psoriatic arthritis from a mechanistic perspective. Nat Rev Rheumatol. 2022;18:311–25.
Garshick MS, Baumer Y, Dey AK, Grattan R, Ng Q, Teague HL, et al. Characterization of PCSK9 in the Blood and Skin of Psoriasis. J Invest Dermatol. 2021;141:308–15.
Katsuki S, K Jha P, Lupieri A, Nakano T, Passos LSA, Rogers MA, et al. Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) Promotes Macrophage Activation via LDL Receptor-Independent Mechanisms. Circ Res. 2022;131:873–89.
Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep. 2018;8:2267.
Candia L, Marquez J, Hernandez C, Zea AH, Espinoza LR. Toll-like receptor-2 expression is upregulated in antigen-presenting cells from patients with psoriatic arthritis: a pathogenic role for innate immunity? J Rheumatol. 2007;34:374–9.
Fitzgerald O, Winchester R. Psoriatic arthritis: from pathogenesis to therapy. Arthritis Res Ther. 2009;11:214.
Van Raemdonck K, Umar S, Palasiewicz K, Romay B, Volkov S, Arami S, et al. TLR7 endogenous ligands remodel glycolytic macrophages and trigger skin-to-joint crosstalk in psoriatic arthritis. Eur J Immunol. 2021;51:714–20.
Diani M, Altomare G, Reali E. T cell responses in psoriasis and psoriatic arthritis. Autoimmun Rev. 2015;14:286–92.
Leijten EFA, van Kempen TS, Boes M, Michels-van Amelsfort JMR, Hijnen D, Hartgring SAY, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. 2015;67:2673–8.
Kim YU, Kee P, Danila D, Teng B-B. A Critical Role of PCSK9 in Mediating IL-17-Producing T Cell Responses in Hyperlipidemia. Immune Netw. 2019;19: e41.
Badimon L, Luquero A, Crespo J, Peña E, Borrell-Pages M. PCSK9 and LRP5 in macrophage lipid internalization and inflammation. Cardiovasc Res. 2021;117:2054–68.
Istvan ES. Structural mechanism for statin inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Am Heart J. 2002;144(6 Suppl):S27-32.
Pirro M, Simental-Mendía LE, Bianconi V, Watts GF, Banach M, Sahebkar A. Effect of Statin Therapy on Arterial Wall Inflammation Based on 18F-FDG PET/CT: A Systematic Review and Meta-Analysis of Interventional Studies. J Clin Med. 2019;8:118.
Faghihi T, Radfar M, Mehrabian Z, Ehsani AH, Rezaei HM. Atorvastatin for the treatment of plaque-type psoriasis. Pharmacotherapy. 2011;31:1045–50.
Cozzani E, Scaparro M, Parodi A. A case of psoriasis worsened by atorvastatin. J Dermatol Case Rep. 2009;3:60–1.
Montanaro S, Lhiaubet-Vallet V, Iesce MI, Previtera L, Miranda MA. A mechanistic study on the phototoxicity of atorvastatin: singlet oxygen generation by a phenanthrene-like photoproduct. Chem Res Toxicol. 2009;22:173–8.
Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–59.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (U22A20162, 32071351, 31900583, 82102604, 81960395), Sanming Project of Medicine in Shenzhen (SZSM201911002), and AOCMF Translational approaches for bone constructs(AOCMF-21-04S, Academic Affairs Office of Sun Yat-sen University (202211583, 202211589 ). Thanks very much for the support of the fund, and thanks also to all the members who have contributed to the idea of the article, data analysis, experimental research, article writing, review, and editing. Special thanks are extended to Dr. Cheng Ruijuan for technical support.
Funding
Sanming Project of Medicine in Shenzen Municipality, SZSM201911002, National Natural Science Foundation of China, U22A20162, 32071351, 31900583, 82102604, 81960395.
Author information
Authors and Affiliations
Contributions
JL. and ZZ had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: JL and JL. Acquisition, analysis, or interpretation of data: CL. Drafting of the manuscript: JL. Critical revision of the manuscript for important intellectual content: JL and JW. Statistical analysis: JZ and JW. Obtained funding: ZZ. Administrative, technical, or material support: FW and HL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflict of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Li, J., Lin, C. et al. Genetically proxied PCSK9 inhibition is associated with reduced psoriatic arthritis risk. Inflamm. Res. 73, 475–484 (2024). https://doi.org/10.1007/s00011-024-01850-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-024-01850-3